
1
MOLECULAR SYMMETRY, GROUP THEORY, & APPLICATIONS
Lecturer: Claire Vallance (CRL office G9, phone 75179, e-mail claire.v[email protected].ac.uk)
These are the lecture notes for the second year general chemistry course named ‘Symmetry I’ in the
course outline. They contain everything in the lecture slides, along with some additional information.
You should, of course, feel free to make your own notes during the lectures if you want to as well. If
anyone would desperately like a copy of the lecture slides, e-mail me at the end of the course and I’ll
send you one (the file is about 2MB in size).
At some point after each lecture and before the next, I STRONGLY recommend that you read the
relevant sections of the lecture handout in order to consolidate the material from the previous lecture
and refresh your memory. Most people (including me!) find group theory quite challenging the first
time they encounter it, and you will probably find it difficult to absorb everything on the first go in
the lectures without doing any additional reading. The good news is that a little extra effort on your
part as we go along should easily prevent you from getting hopelessly lost!
If you have questions at any point, please feel free to ask them either during or after the lectures, or
contact me by e-mail or in the department (contact details above).
Below is a (by no means comprehensive) list of some textbooks you may find useful for the course. If
none of these appeal, have a look in your college library, the Hooke library or the RSL until you find one
that suits you.
Atkins - Physical Chemistry
Atkins - Molecular Quantum Mechanics
Ogden – Introduction to Molecular Symmetry (Oxford Chemistry Primer)
Cotton – Chemical Applications of Group Theory
Davidson – Group Theory for Chemists
Kettle – Symmetry and Structure
Shriver, Atkins and Langford – Inorganic Chemistry
Alan Vincent – Molecular Symmetry and Group Theory (Wiley)
NOTE: A PROBLEM SHEET IS ATTACHED TO THE END OF THIS HANDOUT
2
Contents
1. Introduction
2. Symmetry operations and symmetry elements
3. Symmetry classification of molecules – point groups
4. Symmetry and physical properties
4.1. Polarity
4.2. Chirality
5. Combining symmetry operations: ‘group multiplication’
6. Constructing higher groups from simpler groups
7. Mathematical definition of a group
8. Review of Matrices
8.1. Definitions
8.2. Matrix algebra
8.3 Direct products
8.4. Inverse matrices and determinants
9. Transformation matrices
10. Matrix representations of groups
10.1. Example: a matrix representation of the C
3v
point group (the ammonia molecule)
10.2. Example: a matrix representation of the C
2v
point group (the allyl radical)
11. Properties of matrix representations
11.1. Similarity transforms
11.2. Characters of representations
12. Reduction of representations I
13. Irreducible representations and symmetry species
14. Character tables
15. Reduction of representations II
15.1 General concepts of orthogonality
15.2 Orthogonality relationships in group theory
15.3 Using the LOT to determine the irreps spanned by a basis
16. Symmetry adapted linear combinations
17. Determining whether an integral can be non-zero
18. Bonding in diatomics
19. Bonding in polyatomics - constructing molecular orbitals from SALCs
20. Calculating the orbital energies and expansion coefficients
21. Solving the secular equations
21.1 Matrix formulation of a set of linear equations
21.2 Solving for the orbital energies and expansion coefficients
22. Summary of the steps involved in constructing molecular orbitals
23. A more complicated bonding example – the molecular orbitals of H
2
O
23.1 Matrix representation, characters and SALCs
24. Molecular vibrations
24.1 Molecular degrees of freedom – determining the number of normal vibrational modes
24.2 Determining the symmetries of molecular motions
24.3 Atomic displacements using the 3N Cartesian basis
24.4 Molecular vibrations using internal coordinates
25. Summary of applying group theory to molecular motions
26. Group theory and molecular electronic states
27. Spectroscopy – interaction of atoms and molecules with light
27.1 Electronic transitions in molecules
27.2 Vibrational transitions in molecules
27.3 Raman scattering
28. Summary
29. Appendix A – a few proofs for the mathematically inclined
30. Appendix B – Character tables and direct product tables
Problem sheet

3
1. Introduction
You will already be familiar with the concept of symmetry in an everyday sense. If we say something is
‘symmetrical’, we usually mean it has mirror symmetry, or ‘left-right’ symmetry, and would look the same if viewed
in a mirror. Symmetry is also very important in chemistry. Some molecules are clearly ‘more symmetrical’ than
others, but what consequences does this have, if any?
The aim of this course is to provide a systematic treatment of symmetry in chemical systems within the
mathematical framework known as group theory (the reason for the name will become apparent later on). Once we
have classified the symmetry of a molecule, group theory provides a powerful set of tools that provide us with
considerable insight into many of its chemical and physical properties. Some applications of group theory that will
be covered in this course include:
i) Predicting whether a given molecule will be chiral, or polar.
ii) Examining chemical bonding and visualising molecular orbitals.
iii) Predicting whether a molecule may absorb light of a given polarisation, and which spectroscopic
transitions may be excited if it does.
iv) Investigating the vibrational motions of the molecule.
You may well meet some of these topics again, possibly in more detail, in later courses (notably Symmetry II, and
for the more mathematically inclined amongst you, Supplementary Quantum Mechanics). However, they will be
introduced here to give you a fairly broad introduction to the capabilities and applications of group theory once we
have worked through the basic principles and ‘machinery’ of the theory.
2. Symmetry operations and symmetry elements
A symmetry operation is an action that leaves an object looking the same after it has been carried out. For
example, if we take a molecule of water and rotate it by 180° about an axis passing through the central O atom
(between the two H atoms) it will look the same as before. It will also look the same if we reflect it through
either of two mirror planes, as shown in the figure below.
rotation
(operation)
axis of
symmetry
(element)
reflection
(operation)
mirror plane
(element)
reflection
(operation)
mirror plane
(element)
Each symmetry operation has a corresponding symmetry element, which is the axis, plane, line or point with
respect to which the symmetry operation is carried out. The symmetry element consists of all the points that
stay in the same place when the symmetry operation is performed. In a rotation, the line of points that stay in
the same place constitute a symmetry axis; in a reflection the points that remain unchanged make up a plane of
symmetry.
The symmetry elements that a molecule may possess are:
1. E - the identity. The identity operation consists of doing nothing, and the corresponding symmetry
element is the entire molecule. Every molecule has at least this element.
2. C
n
- an n-fold axis of rotation. Rotation by 360°/n leaves the molecule unchanged. The H
2
O molecule
above has a C
2
axis. Some molecules have more than one C
n
axis, in which case the one with the
highest value of n is called the principal axis. Note that by convention rotations are
counterclockwise about the axis.

4
3. - a plane of symmetry. Reflection in the plane leaves the molecule looking the same. In a molecule
that also has an axis of symmetry, a mirror plane that includes the axis is called a vertical mirror
plane and is labelled
v
, while one perpendicular to the axis is called a horizontal mirror plane and
is labelled
h
. A vertical mirror plane that bisects the angle between two C
2
axes is called a
dihedral mirror plane,
d
.
4. i - a centre of symmetry. Inversion through the centre of symmetry leaves the molecule unchanged.
Inversion consists of passing each point through the centre of inversion and out to the same
distance on the other side of the molecule. An example of a molecule with a centre of inversion is
shown below.
5. S
n
- an n-fold improper rotation axis (also called a rotary-reflection axis). The rotary reflection
operation consists of rotating through an angle 360°/n about the axis, followed by reflecting in a
plane perpendicular to the axis. Note that S
1
is the same as reflection and S
2
is the same as
inversion. The molecule shown above has two S
2
axes.
The identity E and rotations C
n
are symmetry operations that could actually be carried out on a molecule. For this
reason they are called proper symmetry operations. Reflections, inversions and improper rotations can only be
imagined (it is not actually possible to turn a molecule into its mirror image or to invert it without some fairly
drastic rearrangement of chemical bonds) and as such, are termed improper symmetry operations.
A note on axis definitions: Conventionally, when imposing a set of Cartesian axes on a molecule (as we will need to
do later on in the course), the z axis lies along the principal axis of the molecule, the x axis lies in the plane of the
molecule (or in a plane containing the largest number of atoms if the molecule is non-planar), and the y axis makes
up a right handed axis system.
3. Symmetry classification of molecules – point groups
It is only possible for certain combinations of symmetry elements to be present in a molecule (or any other
object). As a result, we may group together molecules that possess the same symmetry elements and classify
molecules according to their symmetry. These groups of symmetry elements are called point groups (due to the
fact that there is at least one point in space that remains unchanged no matter which symmetry operation from
the group is applied). There are two systems of notation for labelling symmetry groups, called the Schoenflies
and Hermann-Mauguin (or International) systems. The symmetry of individual molecules is usually described using
the Schoenflies notation, and we shall be using this notation for the remainder of the course
1
.
Note: Some of the point groups share their names with symmetry operations, so be careful you don’t mix up the
two. It is usually clear from the context which one is being referred to.
The molecular point groups are listed below.
1. C
1
– contains only the identity (a C
1
rotation is a rotation by 360° and is the same as the
identity operation E) e.g. CHDFCl.
1
Though the Hermann-Mauguin system can be used to label point groups, it is usually used in the discussion of crystal symmetry.
In crystals, in addition to the symmetry elements described above, translational symmetry elements are very important.
Translational symmetry operations leave no point unchanged, with the consequence that crystal symmetry is described in terms
of space groups rather than point groups.

5
2. C
i
- contains the identity E and a centre of inversion i.
3. C
S
- contains the identity E and a plane of reflection .
4. C
n
– contains the identity and an n-fold axis of rotation.
5. C
nv
– contains the identity, an n-fold axis of rotation, and n vertical mirror planes
v
.
6. C
nh
- contains the identity, an n-fold axis of rotation, and a horizontal reflection plane
h
(note that
in C
2h
this combination of symmetry elements automatically implies a centre of inversion).
7. D
n
- contains the identity, an n-fold axis of rotation, and n 2-fold rotations about axes perpendicular
to the principal axis.
8. D
nh
- contains the same symmetry elements as D
n
with the addition of a horizontal mirror plane.
9. D
nd
- contains the same symmetry elements as D
n
with the addition of n dihedral mirror planes.
10. S
n
- contains the identity and one S
n
axis. Note that molecules only belong to S
n
if they have not
already been classified in terms of one of the preceding point groups (e.g. S
2
is the same as C
i
,
and a molecule with this symmetry would already have been classified).
The following groups are the cubic groups, which contain more than one principal axis. They separate into the
tetrahedral groups (T
d
, T
h
and T) and the octahedral groups (O and O
h
). The icosahedral group also exists but is
not included below.
11. T
d
– contains all the symmetry elements of a regular tetrahedron, including the identity, 4 C
3
axes, 3
C
2
axes, 6 dihedral mirror planes, and 3 S
4
axes e.g. CH
4
.

6
12. T - as for T
d
but no planes of reflection.
13. T
h
– as for T but contains a centre of inversion.
14. O
h
– the group of the regular octahedron e.g. SF
6
.
15. O - as for O
h
but with no planes of reflection.
The final group is the full rotation group R
3
, which consists of an infinite number of C
n
axes with all possible values
of n and describes the symmetry of a sphere. Atoms (but no molecules) belong to R
3
, and the group has important
applications in atomic quantum mechanics. However, we won’t be treating it any further here.
Once you become more familiar with the symmetry elements and point groups described above, you will find it
quite straightforward to classify a molecule in terms of its point group. In the meantime, the flowchart shown
below provides a step-by-step approach to the problem.
Is the molecule linear?
Does it have a centre
of inversion?
Does it have two or more
C axes with n>2?
n
Does it have a C axis?
n
D
h
C
v
Y
Y
Y
N
N
Y
Does it have a centre
of inversion?
T
d
Does it have a
C axis?
5
I
h
O
h
Y
Y
N
N
START
Does it have a
mirror plane?
C
s
Does it have a centre
of inversion?
C
1
C
i
N
Y
Y
N
N
Are there n C axes
perpendicular to the
principal axis?
2
Is there a horizontal
mirror plane?
Is there a horizontal
mirror plane?
D
nh
Are there n dihedral
mirror planes?
Are there n vertical
mirror planes?
D
nd
D
n
C
nh
C
nv
Y
N
N
N
N
Is there an S
axis?
2n
S
2n
C
n

7
4. Symmetry and physical properties
Carrying out a symmetry operation on a molecule must not change any of its physical properties. It turns out that
this has some interesting consequences, allowing us to predict whether or not a molecule may be chiral or polar on
the basis of its point group.
4.1. Polarity
For a molecule to have a permanent dipole moment, it must have an asymmetric charge distribution. The point
group of the molecule not only determines whether the molecule may have a dipole moment, but also in which
direction(s) it may point.
If a molecule has a C
n
axis with n>1, it cannot have a dipole moment perpendicular to the axis of rotation (for
example, a C
2
rotation would interchange the ends of such a dipole moment and reverse the polarity, which is not
allowed – rotations with higher values of n would also change the direction in which the dipole points). Any dipole
must lie parallel to a C
n
axis.
Also, if the point group of the molecule contains any symmetry operation that would interchange the two ends of
the molecule, such as a
h
mirror plane or a C
2
rotation perpendicular to the principal axis, then there cannot be a
dipole moment along the axis.
The only groups compatible with a dipole moment are C
n
, C
nv
and C
s
. In molecules belonging to C
n
or C
nv
the dipole
must lie along the axis of rotation.
4.2. Chirality
One example of symmetry in chemistry that you will already have come across is found in the isomeric pairs of
molecules called enantiomers. Enantiomers are non-superimposable mirror images of each other, and one
consequence of this symmetrical relationship is that they rotate the plane of polarised light passing through them
in opposite directions. Such molecules are said to be chiral
2
, meaning that they cannot be superimposed on their
mirror image. Formally, the symmetry element that precludes a molecule from being chiral is a rotation-reflection
axis S
n
. Such an axis is often implied by other symmetry elements present in a group. For example, a point group
that has C
n
and
h
as elements will also have S
n
. Similarly, a centre of inversion is equivalent to S
2
. As a rule of
thumb, a molecule definitely cannot have be chiral if it has a centre of inversion or a mirror plane of any type (
h
,
v
or
d
), but if these symmetry elements are absent the molecule should be checked carefully for an S
n
axis
before it is assumed to be chiral.
5. Combining symmetry operations: ‘group multiplication’
Now we will investigate what happens when we apply two symmetry operations in sequence. As an example,
consider the NH
3
molecule, which belongs to the C
3v
point group. Consider what happens if we apply a C
3
rotation
followed by a
v
reflection. We write this combined operation
v
C
3
(when written, symmetry operations operate
on the thing directly to their right, just as operators do in quantum mechanics – we therefore have to work
backwards from right to left from the notation to get the correct order in which the operators are applied). As
we shall soon see, the order in which the operations are applied is important.
1
112 2 2
3
33
v
v
v
'
v
"
C
3
2
The word chiral has its origins in the Greek word for hand (, pronounced ‘cheri’ with a soft ch as in ‘loch’) . A pair of hands
is also a pair of non-superimposable mirror images, and you will often hear chirality referred to as ‘handedness’ for this reason.

8
The combined operation
v
C
3
is equivalent to
v
’’, which is also a symmetry operation of the C
3v
point group. Now
let’s see what happens if we apply the operators in the reverse order i.e. C
3
v
(
v
followed by C
3
).
1
1
1
2 2
2
3 33
v
v
C
3
v
'
v
"
Again, the combined operation C
3
v
is equivalent to another operation of the point group, this time
v
’.
There are two important points that are illustrated by this example:
1. The order in which two operations are applied is important. For two symmetry operations A and B, AB
is not necessarily the same as BA, i.e. symmetry operations do not in general commute. In some groups
the symmetry elements do commute; such groups are said to be Abelian.
2. If two operations from the same point group are applied in sequence, the result will be equivalent to
another operation from the point group. Symmetry operations that are related to each other by other
symmetry operations of the group are said to belong to the same class. In NH
3
, the three mirror
planes
v
,
v
’ and
v
’’ belong to the same class (related to each other through a C
3
rotation), as do the
rotations C
3
+
and C
3
-
(anticlockwise and clockwise rotations about the principal axis, related to each
other by a vertical mirror plane).
The effects of applying two symmetry operations in sequence within a given point group are summarised in group
multiplication tables. As an example, the complete group multiplication table for C
3v
using the symmetry
operations as defined in the figures above is shown below. The operations written along the first row of the table
are carried out first, followed by those written in the first column (note that the table would change if we chose
to name
v
,
v
’ and
v
’’ in some different order).
6. Constructing higher groups from simpler groups
A group that contains a large number of symmetry elements may often be constructed from simpler groups. This
is probably best illustrated using an example. Consider the point groups C
2
and C
S
. C
2
contains the elements E and
C
2
, and has order 2, while C
S
contains E and and also has order 2. We can use these two groups to construct the
group C
2v
by applying the symmetry operations of C
2
and C
S
in sequence.
C
2
operation E E C
2
C
2
C
S
operation E (xz) E (xz)
Result E
v
(xz) C
2
v
’(yz)
Notice that C
2v
has order 4, which is the product of the orders of the two lower-order groups. C
2v
may be
described as a direct product group of C
2
and C
S
. The origin of this name should become obvious when we review
the properties of matrices later on in the course.
C
3v
E
C
3
+
C
3
-
v
v
’
v
’’
E
E
C
3
+
C
3
-
v
v
’
v
’’
C
3
+
C
3
+
C
3
-
E
v
’
v
’’
v
C
3
-
C
3
-
E
C
3
+
v
’
’
v
v
’
v
v
v
’
’
v
’
E
C
3
-
C
3
+
v
’
v
’
v
v
’’
C
3
+
E
C
3
-
v
’’
v
’’
v
’
v
C
3
-
C
3
+
E
9
7. Mathematical definition of a group
Now that we have explored some of the properties of symmetry operations and elements and their behaviour
within point groups, we are ready to introduce the formal mathematical definition of a group.
A mathematical group is defined as a set of elements (g
1
,g
2
,g
3
…) together with a rule for forming combinations
g
i
g
j
. The number of elements h is called the order of the group.
For our purposes, the elements are the symmetry
operations of a molecule and the rule for combining them is the sequential application of symmetry operations
investigated in the previous section. The elements of the group and the rule for combining them must satisfy the
following criteria.
1. The group must include the identity E, for which Eg
i
= g
i
E = g
i
for all the elements of the group.
2. The elements must satisfy the group property that the combination of any pair of elements is also an
element of the group.
3. Each element g
i
must have an inverse g
i
-1
, which is also an element of the group, such that
g
i
g
i
-1
= g
i
-1
g
i
= E (e.g. in C
3v
the inverse of C
3
+
is C
3
-
, the inverse of
v
is
v
; the inverse g
i
-1
‘undoes’
the effect of the symmetry operation g
i
).
4. The rule of combination must be associative i.e. g
i
(g
j
g
k
) = (g
i
g
j
)g
k
The above definition does not require the elements to commute (which would require g
i
g
k
=g
k
g
i
). As we discovered
in the C
3v
example above, in many groups the outcome of consecutive application of two symmetry operations
depends on the order in which the operations are applied. Groups for which the elements do not commute are
called non-Abelian groups; those for which they elements do commute are Abelian.
Group theory is an important area in mathematics, and luckily for chemists the mathematicians have already done
most of the work for us. Along with the formal definition of a group comes a comprehensive mathematical
framework that allows us to carry out a rigorous treatment of symmetry in molecular systems and learn about its
consequences.
Many problems involving operators or operations (such as those found in quantum mechanics or group theory) may
be reformulated in terms of matrices. Any of you who have come across transformation matrices before will know
that symmetry operations such as rotations and reflections may be represented by matrices. It turns out that
the set of matrices representing the symmetry operations in a group obey all the conditions laid out above in the
mathematical definition of a group, and using matrix representations of symmetry operations simplifies carrying
out calculations in group theory. Before we learn how to use matrices in group theory, it will probably be helpful
to review some basic definitions and properties of matrices.
8. Review of Matrices
8.1. Definitions
An nxm matrix is a two dimensional array of numbers with n rows and m columns. The integers n and m are called
the dimensions of the matrix. If n = m then the matrix is square. The numbers in the matrix are known as matrix
elements (or just elements) and are usually given subscripts to signify their position in the matrix e.g. an element
a
ij
would occupy the i
th
row and j
th
column of the matrix. For example:
M =
1 2 3
4 5 6
7 8 9
is a 3x3 matrix with a
11
=1, a
12
=2, a
13
=3, a
21
=4 etc
In a square matrix, diagonal elements are those for which i=j (the numbers 1, 5 and 9 in the above example). Off-
diagonal elements are those for which ij (2, 3, 4, 6, 7, and 8 in the above example). If all the off-diagonal
10
elements are equal to zero then we have a diagonal matrix. We will see later that diagonal matrices are of
considerable importance in group theory.
A unit matrix or identity matrix (usually given the symbol I) is a diagonal matrix in which all the diagonal elements
are equal to 1. A unit matrix acting on another matrix has no effect – it is the same as the identity operation in
group theory and is analogous to multiplying a number by 1 in everyday arithmetic.
The transpose A
T
of a matrix A is the matrix that results from interchanging all the rows and columns. A
symmetric matrix is the same as its transpose (A
T
=A i.e. a
ij
=a
ji
for all values of i and j). The transpose of matrix
M above (which is not symmetric) is
M
T
=
1 4 7
2 5 8
3 6 9
The sum of the diagonal elements in a square matrix is called the trace (or character) of the matrix (for the
above matrix, the trace is = 1 + 5 + 9 = 15). The traces of matrices representing symmetry operations will turn
out to be of great importance in group theory.
A vector is just a special case of a matrix in which one of the dimensions is equal to 1. An nx1 matrix is a column
vector; a 1xm matrix is a row vector. The components of a vector are usually only labelled with one index. A unit
vector has one element equal to 1 and the others equal to zero (it is the same as one row or column of an identity
matrix). We can extend the idea further to say that a single number is a matrix (or vector) of dimension 1x1.
8.2. Matrix algebra
i) Two matrices with the same dimensions may be added or subtracted by adding or subtracting the elements
occupying the same position in each matrix. e.g.
A =
1 0 2
2 2 1
3 2 0
B =
2 0 -2
1 0 1
1 -1 0
A + B =
3 0 0
3 2 2
4 1 0
A – B =
-1 0 4
1 2 0
2 3 0
ii) A matrix may be multiplied by a constant by multiplying each element by the constant.
4B =
8 0 -8
4 0 4
4 -4 0
3A =
3 0 6
6 6 3
9 6 0
iii) Two matrices may be multiplied together provided that the number of columns of the first matrix is the same
as the number of rows of the second matrix i.e. an nxm matrix may be multiplied by an mxl matrix. The
resulting matrix will have dimensions nxl. To find the element a
ij
in the product matrix, we take the dot
product of row i of the first matrix and column j of the second matrix (i.e. we multiply consecutive elements
together from row i of the first matrix and column j of the second matrix and add them together i.e.
c
ij
=
k
a
ik
b
kj
e.g. in the 3x3 matrices A and B used in the above examples, the first element in the product
matrix C = AB is c
11
=a
11
b
11
+a
12
b
21
+a
13
b
31
AB =
1 0 2
2 2 1
3 2 0
2 0 -2
1 0 1
1 -1 0
=
4 -2 -2
7 -1 -2
8 0 -4
An example of a matrix multiplying a vector is
Av =
1 0 2
2 2 1
3 2 0
1
2
3
=
7
9
7

11
Matrix multiplication is not generally commutative, a property that mirrors the behaviour found earlier for
symmetry operations within a point group.
8.3 Direct products
The direct product of two matrices (given the symbol ) is a special type of matrix product that generates a
matrix of higher dimensionality if both matrices have dimension greater than one. The easiest way to
demonstrate how to construct a direct product of two matrices A and B is by an example:
A B =
a
11
a
12
a
21
a
22
b
11
b
12
b
21
b
22
=
a
11
B a
12
B
a
21
B a
22
B
=
a
11
b
11
a
11
b
12
a
12
b
11
a
11
b
12
a
11
b
21
a
11
b
22
a
12
b
21
a
12
b
22
a
21
b
11
a
21
b
12
a
22
b
11
a
22
b
12
a
21
b
21
a
21
b
22
a
22
b
21
a
22
b
22
Though this may seem like a somewhat strange operation to carry out, direct products crop up a great deal in
group theory.
8.4. Inverse matrices and determinants
If two square matrices A and B multiply together to give the identity matrix I (i.e. AB = I) then B is said to be
the inverse of A (written A
-1
). If B is the inverse of A then A is also the inverse of B. Recall that one of the
conditions imposed upon the symmetry operations in a group is that each operation must have an inverse. It
follows by analogy that any matrices we use to represent symmetry elements must also have inverses. It turns
out that a square matrix only has an inverse if its determinant is non-zero. For this reason (and others which will
become apparent later on when we need to solve equations involving matrices) we need to learn a little about
matrix determinants and their properties.
For every square matrix, there is a unique function of all the elements that yields a single number called the
determinant. Initially it probably won’t be particularly obvious why this number should be useful, but matrix
determinants are of great importance both in pure mathematics and in a number of areas of science. Historically,
determinants were actually around before matrices. They arose originally as a property of a system of linear
equations that ‘determined’ whether the system had a unique solution. As we shall see later, when such a system
of equations is recast as a matrix equation this property carries over into the matrix determinant.
There are two different definitions of a determinant, one geometric and one algebraic. In the geometric
interpretation, we consider the numbers across each row of an nxn matrix as coordinates in n-dimensional space.
In a one-dimensional matrix (i.e. a number), there is only one coordinate, and the determinant can be interpreted
as the (signed) length of a vector from the origin to this point. For a 2x2 matrix we have two coordinates in a
plane, and the determinant is the (signed) area of the parallelogram that includes these two points and the origin.
For a 3x3 matrix the determinant is the (signed) volume of the parallelepiped that includes the three points (in
three-dimensional space) defined by the matrix and the origin. This is illustrated below. The idea extends up to
higher dimensions in a similar way. In some sense then, the determinant is therefore related to the size of a
matrix.
(-1)
x
0
-1
1 2
-1 1
(
)
y
x
(1,2)
(-1,1)
1 2 0
-1 1 0
-1 0 1
(
)
x
y
z
(1,2,0)
(-1,1,0)
(-1,0,1)
12
The algebraic definition of the determinant of an nxn matrix is a sum over all the possible products
(permutations) of n elements taken from different rows and columns. The number of terms in the sum is n!, the
number of possible permutations of n values (i.e. 2 for a 2x2 matrix, 6 for a 3x3 matrix etc). Each term in the sum
is given a positive or a negative sign depending on whether the number of permutation inversions in the product is
even or odd. A permutation inversion is just a pair of elements that are out of order when described by their
indices. For example, for a set of four elements (a
1
, a
2
, a
3
, a
4
), the permutation a
1
a
2
a
3
a
4
has all the elements in
their correct order (i.e. in order of increasing index). However, the permutation a
2
a
4
a
1
a
3
contains the permutation
inversions a
2
a
1
, a
4
a
1
, a
4
a
3
.
For example, for a two-dimensional matrix
a
11
a
12
a
21
a
22
where the subscripts label the row and column positions of the elements, there are 2 possible
products/permutations involving elements from different rows and column, a
11
a
22
and a
12
a
21
. In the second term,
there is a permutation inversion involving the column indices 2 and 1 (permutation inversions involving the row and
column indices should be looked for separately) so this term takes a negative sign, and the determinant is a
11
a
22
-
a
12
a
21
.
For a 3x3 matrix
a
11
a
12
a
13
a
21
a
22
a
23
a
31
a
32
a
33
the possible combinations of elements from different rows and columns, together with the sign from the number
of permutations required to put their indices in numerical order are:
a
11
a
22
a
33
(0 inversions)
-a
11
a
23
a
32
(1 inversion – 3>2 in the column indices)
-a
12
a
21
a
33
(1 inversion – 2>1 in the column indices)
a
12
a
23
a
31
(2 inversion2 – 2>1 and 3>1 in the column indices)
a
13
a
21
a
32
(2 inversions – 3>1 and 3>2 in the column indices)
-a
13
a
22
a
31
(3 inversions – 3>2, 3>1 and 2>1 in the column indices)
and the determinant is simply the sum of these terms.
This may all seem a little complicated, but in practice there is a fairly systematic procedure for calculating
determinants. The determinant of a matrix A is usually written det(A) or |A|.
For a 2x2 matrix
A =
a b
c d
det(A) = |A| =
a b
c d
= ad-bc
For a 3x3 matrix
B =
a b c
d e f
g h i
det(B) = a
e f
h i
- b
d f
g i
+ c
d e
g h
13
For a 4x4 matrix
C =
a b c d
e f g h
i j k l
m n o p
det(C) = a
f g h
j k l
n o p
- b
e g h
i k l
m o p
+c
e f h
i j l
m n p
- d
e f g
i j k
m n o
and so on in higher dimensions. Note that the submatrices in the 3x3 example above are just the matrices formed
from the original matrix B that don’t include any elements from the same row or column as the premultiplying
factors from the first row.
Matrix determinants have a number of important properties:
i) The determinant of the identity matrix is 1.
e.g.
1 0
0 1
=
1 0 0
0 1 0
0 0 1
= 1
ii) The determinant of a matrix is the same as the determinant of its transpose i.e. det(A) = det(A
T
)
e.g.
a b
c d
=
a c
b d
iii) The determinant changes sign when any two rows or any two columns are interchanged
e.g.
a b
c d
= -
b a
d c
= -
c d
a b
=
d c
b a
iv) The determinant is zero if any row or column is entirely zero, or if any two rows or columns are equal
or a multiple of one another.
e.g.
1 2
0 0
= 0,
1 2
2 4
= 0
v) The determinant is unchanged by adding any linear combination of rows (or columns) to another row
(or column).
vi) The determinant of the product of two matrices is the same as the product of the determinants of
the two matrices i.e. det(AB) = det(A)det(B).
The requirement that in order for a matrix to have an inverse it must have a non-zero determinant follows from
property vi). As mentioned previously, the product of a matrix and its inverse yields the identity matrix I. We
therefore have:
det(A
-1
A) = det(A
-1
)det(A) = det(I)
det(A
-1
) = det(I)/det(A) = 1/det(A)
It follows that a matrix A can only have an inverse if its determinant is non-zero, otherwise the determinant of
its inverse would be undefined.
9. Transformation matrices
Matrices can be used to map one set of coordinates or functions onto another set. Matrices used for this purpose
are called transformation matrices. In group theory, we can use transformation matrices to carry out the various

14
symmetry operations considered at the beginning of the course. As a simple example, we will investigate the
matrices we would use to carry out some of these symmetry operations on a vector (x,y).
1. The identity operation
The identity operation leaves the vector unchanged, and as you may already suspect, the appropriate matrix is the
identity matrix.
(x,y)
1 0
0 1
= (x,y)
2. Reflection in a plane
The simplest example of a reflection matrix corresponds to reflecting the vector (x,y) in either the x or y axes.
Reflection in the x axis maps y to –y, while reflection in the y axis maps x to -x. The appropriate matrix is very
like the identity matrix but with a change in sign for the appropriate element. Reflection in the x axis transforms
the vector (x,y) to (x,-y), and the appropriate matrix is
(x,y)
1 0
0 -1
= (x,-y)
Reflection in the y axis transforms the vector (x,y)
to (-x,y), and the appropriate matrix is
(x,y)
-1 0
0 1
= (-x,y)
More generally, matrices can be used to represent
reflections in any plane (or line in 2D). For example, reflection in the 45° axis shown below maps (x,y) onto (-y,x).
(x,y)
0 -1
-1 0
= (-y,-x)
3. Rotation about an axis.
In two dimensions, the appropriate matrix to
represent rotation by an angle about the origin is
R() =
cos -sin
sin cos
In three dimensions, rotations about the x, y and z axes acting on a vector (x,y,z) are represented by the
following matrices.
R
x
() =
1 0 0
0 cos -sin
0 sin cos
R
y
() =
cos 0 -sin
0 1 0
sin 0 cos
R
z
() =
cos -sin 0
sin cos 0
0 0 1
10. Matrix representations of groups
We are now ready to integrate what we have just learned about matrices with group theory. The symmetry
operations in a group may be represented by a set of transformation matrices (g), one for each symmetry
element g. Each individual matrix is called a representative of the corresponding symmetry operation, and the
complete set of matrices is called a matrix representation of the group. The matrix representatives act on some
chosen basis set of functions, and the actual matrices making up a given representation will depend on the basis
that has been chosen. The representation is then said to span the chosen basis. In the examples above we were
looking at the effect of some simple transformation matrices on an arbitrary vector (x,y). The basis was
therefore a pair of unit vectors pointing in the x and y directions. In most of the examples we will be considering
in this course, we will use sets of atomic orbitals as basis functions for matrix representations. Don’t worry too
(x,y)
(x,-y)
reflection
in x axis
(x,y)
(-x,y)
reflection
in y axis
(x,y)
(-y,-x)
reflection
in a 45°axis

15
much if these ideas seem a little abstract at the moment – they should become clearer in the next section when
we look at some examples.
Before proceeding any further, we must check that a matrix representation of a group obeys all of the rules set
out in the formal mathematical definition of a group.
1. The first rule is that the group must include the identity operation E (the ‘do nothing’ operation). We
showed above that the matrix representative of the identity operation is simply the identity matrix. As a
consequence, every matrix representation includes the appropriate identity matrix.
2. The second rule is that the combination of any pair of elements must also be an element of the group (the
group property). If we multiply together any two matrix representatives, we should get a new matrix
which is a representative of another symmetry operation of the group. In fact, matrix representatives
multiply together to give new representatives in exactly the same way as symmetry operations combine
according to the group multiplication table. For example, in the C
3v
point group, we showed that the
combined symmetry operation C
3
v
is equivalent to
v
’’. In a matrix representation of the group, if the
matrix representatives of C
3
and
v
are multiplied together, the result will be the representative of
v
’’.
3. The third rule states that every operation must have an inverse, which is also a member of the group.
The combined effect of carrying out an operation and its inverse is the same as the identity operation.
It is fairly easy to show that matrix representatives satisfy this criterion. For example, the inverse of a
reflection is another reflection, identical to the first. In matrix terms we would therefore expect that a
reflection matrix was its own inverse, and that two identical reflection matrices multiplied together
would give the identity matrix. This turns out to be true, and can be verified using any of the reflection
matrices in the examples above. The inverse of a rotation matrix is another rotation matrix
corresponding to a rotation of the opposite sense to the first.
4. The final rule states that the rule of combination of symmetry elements in a group must be associative.
This is automatically satisfied by the rules of matrix multiplication.
10.1. Example: a matrix representation of the C
3v
point group (the ammonia molecule)
The first thing we need to do before we can construct a matrix representation is to choose a basis. For NH
3
, we
will select a basis (s
N
,s
1
,s
2
,s
3
) that consists of the valence s orbitals on the nitrogen and the three hydrogen
atoms. We need to consider what happens to this basis when it is acted on by each of the symmetry operations in
the C
3v
point group, and determine the matrices that would be required to produce the same effect. The basis
set and the symmetry operations in the C
3v
point group are summarised in the figure below.
v
v
'
v
"
C
+
3
C
-
3
s
1
s
2
s
3
s
N
The effects of the symmetry operations on our chosen basis are as follows:
E (s
N
,s
1
,s
2
,s
3
) (s
N
,s
1
,s
2
,s
3
)
C
3
+
(s
N
,s
1
,s
2
,s
3
) (s
N
,s
2
,s
3
,s
1
)
C
3
-
(s
N
,s
1
,s
2
,s
3
) (s
N
,s
3
,s
1
,s
2
)
v
(s
N
,s
1
,s
2
,s
3
) (s
N
,s
1
,s
3
,s
2
)
v
’ (s
N
,s
1
,s
2
,s
3
) (s
N
,s
2
,s
1
,s
3
)
v
’’ (s
N
,s
1
,s
2
,s
3
) (s
N
,s
3
,s
2
,s
1
)
By inspection, the matrices that carry out the same transformations are:

16
(E) (s
N
,s
1
,s
2
,s
3
)
1 0 0 0
0 1 0 0
0 0 1 0
0 0 0 1
= (s
N
,s
1
,s
2
,s
3
)
(C
3
+
) (s
N
,s
1
,s
2
,s
3
)
1 0 0 0
0 0 0 1
0 1 0 0
0 0 1 0
= (s
N
,s
2
,s
3
,s
1
)
(C
3
-
) (s
N
,s
1
,s
2
,s
3
)
1 0 0 0
0 0 1 0
0 0 0 1
0 1 0 0
= (s
N
,s
3
,s
1
,s
2
)
v
) (s
N
,s
1
,s
2
,s
3
)
1 0 0 0
0 1 0 0
0 0 0 1
0 0 1 0
= (s
N
,s
1
,s
3
,s
2
)
v
’) (s
N
,s
1
,s
2
,s
3
)
1 0 0 0
0 0 1 0
0 1 0 0
0 0 0 1
= (s
N
,s
2
,s
1
,s
3
)
(
v
’’) (s
N
,s
1
,s
2
,s
3
)
1 0 0 0
0 0 0 1
0 0 1 0
0 1 0 0
= (s
N
,s
3
,s
2
,s
1
)
These six matrices therefore form a representation for the C
3v
point group in the (s
N
,s
1
,s
2
,s
3
) basis. They
multiply together according to the group multiplication table and satisfy all the requirements for a mathematical
group.
Note: We have written the vectors representing our basis as row vectors. This is important. If we had written
them as column vectors, the corresponding transformation matrices would be the transposes of the matrices
above, and would not reproduce the group multiplication table (try it as an exercise if you need to convince
yourself).
10.2. Example: a matrix representation of the C
2v
point group (the allyl radical)
In this example, we’ll take as our basis a p orbital on each carbon atom (p
1
,p
2
,p
3
).
p
1
p
2
p
3
Note that the p orbitals are perpendicular to the plane of the carbon atoms (this may seem obvious, but if you’re
visualising the basis incorrectly it will shortly cause you a not inconsiderable amount of confusion). The symmetry
operations in the C
2v
point group, and their effect on the three p orbitals, are as follows:
E (p
1
,p
2
,p
3
) (p
1
,p
2
,p
3
)
C
2
(p
1
,p
2
,p
3
) (-p
3
,-p
2
,-p
1
)
v
(p
1
,p
2
,p
3
) (-p
1
,-p
2
,-p
3
)
v
’ (p
1
,p
2
,p
3
) (p
3
,p
2
,p
1
)
The matrices that carry out the transformation are
17
(E) (p
1
,p
2
,p
3
)
1 0 0
0 1 0
0 0 1
= (p
1
,p
2
,p
3
)
(C
2
) (p
1
,p
2
,p
3
)
0 0 -1
0 -1 0
-1 0 0
= (-p
3
,-p
2
,-p
1
)
(
v
) (p
1
,p
2
,p
3
)
-1 0 0
0 -1 0
0 0 -1
= (-p
1
,-p
2
,-p
3
)
(
v
’) (p
1
,p
2
,p
3
)
0 0 1
0 1 0
1 0 0
= (p
3
,p
1
,p
2
)
11. Properties of matrix representations
Now that we’ve learnt how to create a matrix representation of a point group within a given basis, we will move on
to look at some of the properties that make these representations so powerful in the treatment of molecular
symmetry.
11.1. Similarity transforms
Suppose we have a basis set (x
1
,x
2
,x
3
,…x
n
), and we have determined the matrix representatives for the basis in a
given point group. There is nothing particularly special about the basis set we have chosen, and we could equally
well have used any set of linear combinations of the original functions (provided the combinations were linearly
independent). The matrix representatives for the two basis sets will certainly be different, but we would expect
them to be related to each other in some way. As we shall show shortly, they are in fact related by a similarity
transform. It will be far from obvious at this point why we would want to carry out such a transformation, but
similarity transforms will become important later on when we use group theory to choose an optimal basis set with
which to generate molecular orbitals.
Consider a basis set (x
1
’,x
2
’,x
3
’,…x
n
’), in which each basis function x
i
’ is a linear combination of our original basis
(x
1
,x
2
,x
3
,…x
n
).
x
j
’ =
i
x
i
c
ji
= x
1
c
j1
+ x
2
c
j2
+ …
The c
ji
appearing in the sum are coefficients; c
ji
is the coefficient multiplying the original basis function x
i
in the
new linear combination basis function x
j
’. We could also represent this transformation in terms of a matrix
equation x’ = xC:
(x
1
’,x
2
’,…x
n
’) = (x
1
,x
2
,…x
n
)
c
11
c
12
… c
1n
c
21
c
22
… c
2n
… … … …
c
n1
c
n2
… c
nn
Now we look at what happens when we apply a symmetry operation g to our two basis sets. If (g) and ’(g) are
matrix representatives of the symmetry operation in the x and x’ bases, then we have:
gx’ = x’’(g)
gxC = xC’(g) since x’ = xC
gx = xC’(g)C
-1
multiplying on the right by C
-1
and using CC
-1
= I
= x (g)

18
We can therefore identify the similarity transform relating (g), the matrix representative in our original basis,
to ’(g), the representative in the transformed basis. The transform depends only on the matrix of coefficients
used to transform the basis functions.
(g) = C’(g)C
-1
Also ’(g) = C
-1
(g)C
11.2. Characters of representations
The trace of a matrix representative (g) is usually referred to as the character of the representation under the
symmetry operation g. We will soon come to see that the characters of a matrix representation are often more
useful than the matrix representatives themselves. Characters have several important properties.
1. The character of a symmetry operation is invariant under a similarity transform
2. Symmetry operations belonging to the same class have the same character in a given representation. Note
that the character for a given class may be different in different representations, and that more than one class
may have the same character.
Proofs of the above two statements are given in the Appendix.
12. Reduction of representations I
Let us now go back and look at the C
3v
representation we derived in 10.1 in more detail. If we look at the matrices
carefully we see that they all take the same block diagonal form (a square matrix is said to be block diagonal if all
the elements are zero except for a set of submatrices lying along the diagonal).
(E)
(E) = 4
(C
)
3
+
(C ) = 1
3
+
(C
)
3
-
(C ) = 1
3
-
(
v
)
) = 2(
v
(
’)
v
( ’) = 2
v
(
’’)
v
( ’’) = 2
v
1 0 0 0
0 1 0 0
0 0 1 0
0 0 0 1
1 0 0 0
0 0 0 1
0 1 0 0
0 0 1 0
1 0 0 0
0 0 1 0
0 0 0 1
0 1 0 0
1 0 0 0
0 1 0 0
0 0 0 1
0 0 1 0
1 0 0 0
0 0 1 0
0 1 0 0
0 0 0 1
1 0 0 0
0 0 0 1
0 0 1 0
0 1 0 0
A block diagonal matrix can be written as the direct sum of the matrices that lie along the diagonal. In the case
of the C
3v
matrix representation, each of the matrix representatives may be written as the direct sum of a 1x1
matrix and a 3x3 matrix.
(4)
(g) =
(1)
(g)
(3)
(g)
in which the bracketed superscripts denote the dimensionality of the matrices. Note that a direct sum is very
different from ordinary matrix addition since it produces a matrix of higher dimensionality. A direct sum of two
matrices of orders n and m is performed by placing the matrices to be summed along the diagonal of a matrix of
order n+m and filling in the remaining elements with zeroes.
The reason why this result is useful in group theory is that the two sets of matrices
(1)
(g) and
(3)
(g) also satisfy
all of the requirements for a matrix representation. Each set contains the identity and an inverse for each
member, and the members multiply together associatively according to the group multiplication table
3
. Recall that
the basis for the original four-dimensional representation had the s orbitals (s
N
,s
1
,s
2
,s
3
) of ammonia as its basis.
The first set of reduced matrices,
(1)
(g), forms a one-dimensional representation with (s
N
) as its basis. The
second set,
(3)
(g) forms a three-dimensional representation with the basis (s
1
,s
2
,s
3
). Separation of the original
representation into representations of lower dimensionality is called reduction of the representation. The two
reduced representations are shown below.
3
The 1x1 representation in which all of the elements are equal to 1 is sometimes called the unfaithful representation, since it
satisfies the group properties in a fairly trivial way without telling us much about the symmetry properties of the group.

19
g E C
3
+
C
3
-
v
v
’
v
’’
1D representation spanned
(1)
(g) (1) (1) (1) (1) (1) (1) by (s
N
)
(3)
(g)
1 0 0
0 1 0
0 0 1
0 1 0
0 0 1
1 0 0
0 0 1
1 0 0
0 1 0
1 0 0
0 0 1
0 1 0
0 1 0
1 0 0
0 0 1
0 0 1
0 1 0
1 0 0
3D representation spanned
by (s
1
,s
2
,s
3
)
The logical next step is to investigate whether or not the three dimensional representation
(3)
(g) can be reduced
any further. As it stands, the matrices making up this representation are not in block diagonal form (some of you
may have noted that the matrices representing E and
v
are block diagonal, but in order for a representation to be
reducible all of the matrix representatives must be in the same block diagonal form) so the representation is not
reducible. However, we can carry out a similarity transformation (see 10.1) to a new representation spanned by a
new set of basis functions (made up of linear combinations of (s
1
,s
2
,s
3
)), which is reducible. In this case, the
appropriate (normalised) linear combinations to use as our new basis functions are
s
1
’ =
1
3
(s
1
+ s
2
+ s
3
)
s
2
’ =
1
6
(2s
1
– s
2
–s
3
)
s
3
’ =
1
2
(s
2
– s
3
)
or in matrix form
(s
1
’,s
2
’,s
3
’) = (s
1
,s
2
,s
3
)
1/ 3 2/ 6 0
1/ 3 -1/ 6 1/ 2
1/ 3 -1/ 6 -1/ 2
x’ = x C
The matrices in the new representation are found from ’(g) = C
-1
(g)C to be
E C
3
+
C
3
-
v
v
’
v
’’
(3)
’(g)
1 0 0
0 1 0
0 0 1
1 0 0
0 -1/2 3/2
0 - 3/2 -1/2
1 0 0
0 -1/2 - 3/2
0 3/2 -1/2
1 0 0
0 1 0
0 0 -1
1 0 0
0 -1/2 3/2
0 3/2 1/2
1 0 0
0 -1/2 - 3/2
0 - 3/2 1/2
We see that each matrix is now in block diagonal form, and the representation may be reduced into the direct sum
of a 1x1 representation spanned by (s
1
’) and a 2x2 representation spanned by (s
2
’,s
3
’). The complete set of
reduced representations obtained from the original 4D representation is:
E C
3
+
C
3
-
v
v
’
v
’’
(1) (1) (1) (1) (1) (1) 1D representation spanned by (s
N
)
(1) (1) (1) (1) (1) (1) 1D representation spanned by (s
1
’)
1 0
0 1
-1/2 3/2
- 3/2 -1/2
-1/2 - 3/2
3/2 -1/2
1 0
0 -1
-1/2 3/2
3/2 1/2
-1/2 - 3/2
- 3/2 1/2
2D representation spanned
by (s
2
',s
3
')
This is as far as we can go in reducing this representation. None of the three representations above can be
reduced any further, and they are therefore called irreducible representations, or ‘irreps’, of the point group.
Formally, a representation is an irrep if there is no similarity transform that can simultaneously convert all of the
representatives into block diagonal form. The linear combination of basis functions that converts a matrix
representation into block diagonal form, allowing reduction of the representation, is called a symmetry adapted
linear combination.
s
N
s ’
1
s ’
2
s ’
3

20
13. Irreducible representations and symmetry species
The two one-dimensional irreps spanned by s
N
and s
1
’ are seen to be identical. This means that s
N
and s
1
’ have the
‘same symmetry’, transforming in the same way under all of the symmetry operations of the point group and
forming bases for the same matrix representation. As such, they are said to belong to the same symmetry
species. There are a limited number of ways in which an arbitrary function can transform under the symmetry
operations of a group, giving rise to a limited number of symmetry species. Any function that forms a basis for a
matrix representation of a group must transform as one of the symmetry species of the group. The irreps of a
point group are labelled according to their symmetry species as follows:
i) 1D representations are labelled A or B, depending on whether they are symmetric (character +1) or
antisymmetric (character –1) under rotation about the principal axis.
ii) 2D representations are labelled E, 3D representations are labelled T.
iii) In groups containing a centre of inversion, g and u labels (from the German gerade and ungerade, meaning
symmetric and antisymmetric) denote the character of the irrep under inversion (+1 for g, -1 for u)
iv) In groups with a horizontal mirror plane but no centre of inversion, the irreps are given prime and double
prime labels to denote whether they are symmetric (character +1) or antisymmetric (character –1) under
reflection in the plane.
v) If further distinction between irreps is required, subscripts 1 and 2 are used to denote the character with
respect to a C
2
rotation perpendicular to the principal axis, or with respect to a vertical reflection if there
are no C
2
rotations.
The 1D irrep in the C
3v
point group is symmetric (has character +1) under all the symmetry operations of the
group. It therefore belongs to the irrep A
1
. The 2D irrep has character 2 under the identity operation, -1 under
rotation, and 0 under reflection, and belongs to the irrep E.
Sometimes there is confusion over the relationship between a function f and its irreducible representation, but it
is quite important that you understand the connection. There are several different ways of stating the
relationship. For example, the following statements all mean the same thing:
“f has A
2
symmetry”
“f transforms as A
2
”
“f has the same symmetry as A
2
”
“f forms a basis for the A
2
irrep”
The most important point to understand is that every function transforms as one of the irreps of a point group.
In the case of one-dimensional irreps there is a one-to-one correspondence between the function and its irrep. In
the case of two-dimensional irreps, a pair of degenerate functions will transform jointly as the 2D irrep, and so
on. The same function may transform as a different irrep in different point groups. For example, a p
z
orbital on
an atom in a tetrahedral environment (e.g. a p
z
orbital on the C atom in CH
4
) transforms as T
2
(along with the two
other p orbitals), while a p
z
orbital lying along the rotation axis of a C
3v
molecule (such as the p
z
orbital on the N
atom in NH
3
) transforms as A
1
.
14. Character tables
A character table summarises the behaviour of all of the possible irreps of a group under each of the symmetry
operations of the group. The character table for C
3v
is shown below.
C
3v
,3m E 2C
3
3
v
h=6
A
1
1 1 1 z, z
2
, x
2
+y
2
A
2
1
1
-
1
R
z
E 2 -1 0 (x,y), (xy,x
2
+y
2
), (xz,yz), (R
x
,R
y
)

21
The various sections of the table are as follows:
i) The first element in the table gives the name of the point group, usually in both Schoenflies (C
3v
) and
Hermann-Mauguin (3m) notation.
ii) Along the first row are the symmetry operations of the group, E, 2C
3
and 3
v
, followed by the order h
of the group. Because operations in the same class have the same character, symmetry operations are
grouped into classes in the character table and not listed separately.
iii) In the first column are the irreps of the group. In C
3v
the irreps are A
1
, A
2
and E (the representation
we considered above spans 2A
1
+ E).
iv) The characters of the irreps under each symmetry operation are given in the bulk of the table.
v) The final column of the table lists a number of functions that transform as the various irreps of the
group. These are the Cartesian axes (x,y,z) the Cartesian products (z
2
, x
2
+y
2
, xy, xz, yz) and the
rotations (R
x
,R
y
,R
z
).
The functions listed in the final column of the table are important in many chemical applications of group theory,
particularly in spectroscopy. For example, by looking at the transformation properties of x, y and z (sometimes
given in character tables as T
x
, T
y
, T
z
) we can discover the symmetry of translations along the x, y, and z axes.
Similarly, R
x
, R
y
and R
z
represent rotations about the three Cartesian axes. As we shall see later, the
transformation properties of x, y, and z can also be used to determine whether or not a molecule can absorb a
photon of x-, y- or z-polarised light and undergo a spectroscopic transition. The Cartesian products play a similar
role in determining selection rules for Raman transitions, which involve two photons.
Character tables for common point groups are given in Appendix B.
Note 1: A simple way to determine the characters of a representation.
In many applications of group theory, we only need to know the characters of the representative matrices, rather
than the matrices themselves. Luckily, when each basis function transforms as a 1D irrep (which is true in many
cases of interest) there is a simple shortcut to determining the characters without having to construct the entire
matrix representation. All we have to do is to look at the way the individual basis functions transform under each
symmetry operation. For a given operation, step through the basis functions as follows:
i) Add 1 to the character if the basis function is unchanged by the symmetry operation (i.e. the basis
function is mapped onto itself);
ii) Add –1 to the character if the basis function changes sign under the symmetry operation (i.e the basis
function is mapped onto minus itself);
iii) Add 0 to the character if the basis function moves when the symmetry operation is applied (i.e the
basis function is mapped onto something different from itself).
Try this for the s orbital basis we have been using for the C
3v
group. You should find you get the same characters
as we obtained from the traces of the matrix representatives.
We can also work out the characters fairly easily when two basis functions transform together as a 2D irrep. For
example, in the C
3v
point group x and y axes transform together as E. If we carry out a rotation about z by an
angle , our x and y axes are transformed onto new axes x’ and y’. However, the new axes can each be written as a
linear combination of our original x and y axes. Using the rotation matrices introduced in Section 9, we see that:
x’ = cos x + sin y
y’ = -sin x + cos y
For one-dimensional irreps we asked if a basis function/axis was mapped onto itself, minus itself, or something
different. For two-dimensional irreps we need to ask how much of the ‘old’ axis is contained in the new one. From
the above we see that the x’ axis contains a contribution cos from the x axis, and the y’ axis contains a

22
contribution cos from the y axis. The characters of the x and y axes under a rotation through are therefore
cos, and the overall character of the E irrep is therefore cos + cos = 2cos. For a C
3
rotation through 120
degrees, the character of the E irrep is therefore 2cos120° = -1.
In general, when an axis is rotated by an angle
by a symmetry operation, its contribution to the character for
that operation is cos
.
Note 2: Irreps with complex characters
In many cases (see Appendix B), the characters for rotations C
n
and improper rotations S
n
are complex numbers,
usually expressed in terms of the quantity = exp(2i/n). It is fairly straightforward to reconcile this with the
fact that in chemistry we are generally using group theory to investigate physical problems in which all quantities
are real. It turns out that whenever our basis spans an irrep whose characters are complex, it will also span a
second irrep whose characters are the complex conjugates of the first irrep i.e. complex irreps occur in pairs.
According to the strict mathematics of group theory, each irrep in the pair should be considered as a separate
representation. However, when applying such irreps in physical problems, we add the characters for the two
irreps together to get a single irrep whose characters are real.
As an example, the ‘correct’ character table for the group C
3
takes the form:
C
3
E C
3
C
3
2
A 1 1 1
E
{
1
1
}
Where = exp(2i/3). However, as chemists we would usually combine the two parts of the E irrep to give:
C
3
E
C
3
C
3
2
A
1
1
1
E 2 -1 -1
15. Reduction of representations II
By making maximum use of molecular symmetry, we often greatly simplify problems involving molecular properties.
For example, the formation of chemical bonds is strongly dependent on the atomic orbitals involved having the
correct symmetries. In order to make full use of group theory in the applications we will be considering, we need
to develop a little more ‘machinery’. Specifically, given a basis set (of atomic orbitals, for example) we need to
find out:
1. How to determine the irreps spanned by the basis functions
2. How to construct linear combinations of the original basis functions that transform as a given
irrep/symmetry species.
It turns out that both of these problems can be solved using something called the ‘Great Orthogonality Theorem’
(GOT for short). The GOT summarises a number of orthogonality relationships implicit in matrix representations
of symmetry groups, and may be derived in a somewhat qualitative fashion by considering these relationships in
turn.
Note: Some of you might find the next section a little hard going. In it, we will derive two important expressions
that we can use to achieve the two goals we have set out above. It is not important that you understand every
step in these derivations; they have mainly been included just so you can see where the equations come from.
However, you will need to understand how to use the results. Hopefully you won’t find this too difficult once we’ve
worked through a few examples.
15.1 General concepts of orthogonality

23
You are probably already familiar with the geometric concept of orthogonality. Two vectors are orthogonal if
their dot product (i.e. the projection of one vector onto the other) is zero. An example of a pair of orthogonal
vectors is provded by the x and y Cartesian unit vectors.
x.y = 0
A consequence of the orthogonality of x and y is that any general vector in the xy plane may be written as a linear
combination of these two basis vectors.
r = ax + by
Mathematical functions may also be orthogonal. Two functions, f
1
(x) and f
2
(x), are defined to be orthogonal if the
integral over their product is equal to zero i.e.
f
1
(x) f
2
(x) dx =
12
. This simply means that there must be ‘no
overlap’ between orthogonal functions, which is the same as the orthogonality requirement for vectors, above. In
the same way as for vectors, any general function may be written as a linear combination of a suitably chosen set
of orthogonal basis functions. For example, the Legendre polynomials P
n
(x) form an orthogonal basis set for
functions of one variable x.
f(x) =
n
c
n
P
n
(x)
15.2 Orthogonality relationships in group theory
The irreps of a point group satisfy a number of orthogonality relationships:
1. If corresponding matrix elements in all of the matrix representatives of an irrep are squared and added
together, the result is equal to the order of the group divided by the dimensionality of the irrep. i.e.
g
k
(g)
ij
k
(g)
ij
=
h
d
k
(15.2.1)
where k labels the irrep, i and j label the row and column position within the irrep, h is the order of the group,
and d
k
is the order of the irrep.
e.g. The order of the group C
3v
is 6. If we apply the above operation to the first element in the 2x2 (E) irrep
derived in Section 12, the result should be equal to h/d
k
= 6/2 = 3. Carrying out this operation gives:
(1)
2
+ (-½)
2
+ (-½)
2
+ (1)
2
+ (-½)
2
+ (-½)
2
= 1 + ¼ + ¼ + 1 + ¼ + ¼ = 3
2. If instead of summing the squares of matrix elements in an irrep, we sum the product of two different
elements from within each matrix, the result is equal to zero. i.e.
g
k
(g)
ij
k
(g)
i'j'
= 0 (15.2.2)
where i i’ and/or j j’.
e.g. if we perform this operation using the two elements in the first row of the 2D irrep used in 1., we get:
y
x
y
r
x

24
(1)(0) + (-½)(
3
2
) + (-½)(-
3
2
) + (1)(0) + (-½)(
3
2
) + (-½)(-
3
2
) = 0 +
3
4
-
3
4
+ 0 –
3
4
+
3
4
= 0
3. If we sum the product of two elements from the matrices of two different irreps k and m, the result is equal
to zero. i.e.
g
k
(g)
ij
m
(g)
i'j'
= 0 (15.2.3)
where there is now no restriction on the values of the indices i,j,i’,j’ (apart from the rather obvious restriction
that they must be less than or equal to the dimensions of the irrep).
e.g. Performing this operation on the first elements of the A
1
and E irreps we derived for C
3v
gives:
(1)(1) + (1)(-½) + (1)(-½) + (1)(1) + (1)(-½) + (1)(-½) = 1 – ½ - ½ + 1 – ½ - ½ = 0
We can combine these three results into one general equation, the Great Orthogonality Theorem
4
.
g
k
(g)
ij
m
(g)
i'j'
=
h
d
k
d
m
km
ii'
jj'
(15.2.4)
For most applications we don’t actually need the full Great Orthogonality Theorem. A little mathematical trickery
transforms Equation (15.2.4) into the ‘Little Orthogonality Theorem’ (or LOT), which is expressed in terms of the
characters of the irreps rather than the irreps themselves.
g
k
(g)
m
(g) = h
km
(15.2.5)
Since the characters for two symmetry operations in the same class are the same, we can also rewrite the sum
over symmetry operations as a sum over classes.
C
n
C
k
(C)
m
(C) = h
km
(15.2.6)
where n
C
is the number of symmetry operations in class C.
In all of the examples we’ve considered so far, the characters have been real. However, this is not necessarily
true for all point groups, so to make the above equations completely general we need to include the possibility of
imaginary characters. In this case we have:
C
n
C
k
*
(C)
m
(C) = h
km
(15.2.7)
where
k
*
(C) is the complex conjugate of
k
(C). Equation (15.2.7) is of course identical to (15.2.6) when all the
characters are real.
15.3 Using the LOT to determine the irreps spanned by a basis
In Section 12 we discovered that we can often carry out a similarity transform on a general matrix representation
so that all the representatives end up in the same block diagonal form. When this is possible, each set of
submatrices also forms a valid matrix representation of the group. If none of the submatrices can be reduced
further by carrying out another similarity transform, they are said to form an irreducible representation of the
point group. An important property of matrix representatives (see Section 11.2) is that their character is
invariant under a similarity transform. This means that the character of the original representatives must be
equal to the sum of the characters of the irreps into which the representation is reduced. e.g. if we consider the
representative for the C
3
-
symmetry operation in our NH
3
example, we have:
4
The
ij
appearing in Equation 15.2.4 are called Dirac delta functions. They are equal to 1 if i = j and 0 otherwise.

25
1 0 0 0
0 0 0 1
0 1 0 0
0 0 1 0
similarity transform
1 0 0 0
0 1 0 0
0 0 -1/2 - 3/2
0 0 3/2 -1/2
= ( 1 ) ( 1 )
-1/2 - 3/2
3/2 -1/2
= 1 = 1 = 1 + 1 + -1 = 1
It follows that we can write the characters for a general representation (g) in terms of the characters of the
irreps
k
(g) into which it can be reduced.
(g) =
k
a
k
k
(g) (15.3.1)
where the coefficients a
k
in the sum are the number of times each irrep appears in the representation. This
means that in order to determine the irreps spanned by a given basis. all we have to do is determine the
coefficients a
k
in the above equation. This is where the Little Orthogonality Theorem comes in handy. If we take
the LOT in the form of Equation 15.2.5, and multiply each side through by a
k
, we get
g
a
k
k
(g)
m
(g) = h a
k
km
(15.3.2)
Summing both sides of the above equation over k gives
g
k
a
k
k
(g)
m
(g) = h
k
a
k
km
We can use Equation (15.3.1) to simplify the left hand side of this equation. Also, the sum on the right hand side
reduces to a
m
because
km
is only non-zero (and equal to 1) when k=m
g
(g)
m
(g) = h a
m
Dividing both sides through by h (the order of the group), gives us an expression for the coefficients a
m
in terms
of the characters (g) of the original representation and the characters
m
(g) of the m
th
irrep.
a
m
=
1
h
g
(g)
m
(g) (15.3.3)
We can of course write this as a sum over classes rather than a sum over symmetry operations.
a
m
=
1
h
C
n
C
(g)
m
(g) (15.3.4)
As an example, in Section 12 we showed that the matrix representatives we derived for the C
3v
group could be
reduced into two irreps of A
1
symmetry and one of E symmetry. i.e. = 2A
1
+ E. We could have obtained the same
result using Equation (15.3.4). The characters for our original representation and for the irreps of the C
3v
point
group (A
1
, A
2
and E) are given in the table below.
C
3v
E 2C
3
3
v
4
1
2
(A
1
)
1 1 1
(A
2
)
1 1 -1
(E)
2 -1 0
From (15.3.4), the number of times each irrep occurs for our chosen basis (s
N
,s
1
,s
2
,s
3
) is therefore
a(A
1
) =
1
6
( 1x4x1 + 2x1x1 + 3x2x1 ) = 2
a(A
2
) =
1
6
( 1x4x1 + 2x1x1 + 3x2x-1 ) = 0

26
a(E) =
1
6
( 1x4x2 + 2x1x-1 + 3x2x0 ) = 1
i.e. Our basis is spanned by 2A
1
+ E, as we found before.
16. Symmetry adapted linear combinations
Once we know the irreps spanned by an arbitrary basis set, we can work out the appropriate linear combinations of
basis functions that transform the matrix representatives of our original representation into block diagonal form
(i.e. the symmetry adapted linear combinations). Each of the SALCs transforms as one of the irreps of the
reduced representation. We have already seen this in our NH
3
example. The two linear combinations of A
1
symmetry were s
N
and s
1
+ s
2
+ s
3
, both of which are symmetric under all the symmetry operations of the point
group. We also chose another pair of functions, 2s
1
– s
2
– s
3
and s
2
– s
3
, which together transform as the
symmetry species E.
To find the appropriate SALCs to reduce a matrix representation, we use projection operators. You will be
familiar with the idea of operators from quantum mechanics. The operators we will be using here are not quantum
mechanical operators, but the basic principle is the same. The projection operator to generate a SALC that
transforms as an irrep k is
g
k
(g) g. Each term in the sum means ‘apply the symmetry operation g and then
multiply by the character of g in irrep k’. Applying this operator to each of our original basis functions in turn will
generate a complete set of SALCs. i.e. to transform a basis function f
i
into a SALC f
i
’, we use
f
i
’ =
g
k
(g) g f
i
(16.1)
The way in which this operation is carried out will become much more clear if we work through an example. We
can break down the above equation into a fairly straightforward ‘recipe’ for generating SALCs:
1. Make a table with columns labelled by the basis functions and rows labelled by the symmetry
operations of the molecular point group. In the columns, show the effect of the symmetry operations
on the basis functions (this is the g f
i
part of Equation (16.1)).
2. For each irrep in turn, multiply each member of the table by the character of the appropriate
symmetry operation (we now have
k
(g) g f
i
for each operation). Summing over the columns (symmetry
operations) generates all the SALCs that transform as the chosen irrep.
3. Normalise the SALCs.
Earlier (see Section 10), we worked out the effect of all the symmetry operations in the C
3v
point group on the
(s
N
,s
1
,s
2
,s
3
) basis.
E (s
N
,s
1
,s
2
,s
3
) (s
N
,s
1
,s
2
,s
3
)
C
3
+
(s
N
,s
1
,s
2
,s
3
) (s
N
,s
2
,s
3
,s
1
)
C
3
-
(s
N
,s
1
,s
2
,s
3
) (s
N
,s
3
,s
1
,s
2
)
v
(s
N
,s
1
,s
2
,s
3
) (s
N
,s
1
,s
3
,s
2
)
v
’ (s
N
,s
1
,s
2
,s
3
) (s
N
,s
2
,s
1
,s
3
)
v
’’ (s
N
,s
1
,s
2
,s
3
) (s
N
,s
3
,s
2
,s
1
)
This is all we need to construct the table described in 1. above.
s
N
s
1
s
2
s
3
E
s
N
s
1
s
2
s
3
C
3
+
s
N
s
2
s
3
s
1
C
3
-
s
N
s
3
s
1
s
2
v
s
N
s
1
s
3
s
2
v
’
s
N
s
2
s
1
s
3
v
’’ s
N
s
3
s
2
s
1

27
To determine the SALCs of A
1
symmetry, we multiply the table through by the characters of the A
1
irrep (all of
which take the value 1). Summing the columns gives
s
N
+ s
N
+ s
N
+ s
N
+ s
N
+ s
N
= 6s
N
s
1
+ s
2
+ s
3
+ s
1
+ s
2
+ s
3
= 2(s
1
+ s
2
+ s
3
)
s
2
+ s
3
+ s
1
+ s
3
+ s
1
+ s
2
= 2(s
1
+ s
2
+ s
3
)
s
3
+ s
1
+ s
2
+ s
2
+ s
3
+ s
1
= 2(s
1
+ s
2
+ s
3
)
Apart from a constant factor (which doesn’t affect the functional form and therefore doesn’t affect the
symmetry properties), these are the same as the combinations we determined earlier. Normalising gives us two
SALCs of A
1
symmetry.
1
= s
N
2
=
1
3
(s
1
+ s
2
+ s
3
)
We now move on to determine the SALCs of E symmetry. Multiplying the table above by the appropriate
characters for the E irrep gives
s
N
s
1
s
2
s
3
E 2s
N
2s
1
2s
2
2s
3
C
3
+
-
s
N
-
s
2
-
s
3
-
s
1
C
3
-
-s
N
-s
3
-s
1
-s
2
v
0
0
0
0
v
’ 0 0 0 0
v
’’
0
0
0
0
Summing the columns yields
2s
N
– s
N
– s
N
= 0
2s
1
– s
2
– s
3
2s
2
– s
3
– s
1
2s
3
– s
1
– s
2
We therefore get three SALCs from this procedure. This is a problem, since the number of SALCs must match
the dimensionality of the irrep, in this case two. Put another way, we should end up with four SALCs in total to
match our original number of basis functions. Added to our two SALCs of A
1
symmetry, three SALCs of E
symmetry would give us five in total.
The resolution to our problem lies in the fact that the three SALCs above are not linearly independent. Any one
of them can be written as a linear combination of the other two e.g. (2s
1
–s
2
–s
3
) = -(2s
2
-s
3
-s
1
) – (2s
3
-s
1
-s
2
). To solve
the problem, we can either throw away one of the SALCs, or better, make two linear combinations of the three
SALCs that are orthogonal to each other.
5
e.g. if we take 2s
1
– s
2
– s
3
as one of our SALCs and find an orthogonal
combination of the other two (which turns out to be their difference), we have (after normalisation)
3
=
1
6
(2s
1
– s
2
– s
3
)
4
=
1
2
(s
2
– s
3
)
These are the same linear combinations used in Section 12.
5
If we write the coefficients of s
1
, s
2
and s
3
for each SALC as a vector (a
1
,a
2
,a
3
), then when two SALCs are orthogonal, the dot
product of their coefficient vectors (a
1
,a
2
,a
3
) (b
1
,b
2
,b
3
) = a
1
b
1
+ a
2
b
2
+ a
3
b
3
is equal to zero).

28
We now have all the machinery we need to apply group theory to a range of chemical problems. In our first
application, we will learn how to use molecular symmetry and group theory to help us understand chemical bonding.
17. Determining whether an integral can be non-zero
As we continue with this course, we will discover that there are many times when we would like to know whether a
particular integral is necessarily zero, or whether there is a chance that it may be non-zero. We can often use
group theory to differentiate these two cases.
You will have already used symmetry properties of functions to determine whether or not a one-dimensional
integral is zero. For example, sin(x) is an ‘odd’ function (antisymmetric with respect to reflection through the
origin), and it follows from this that
-∞
∞
sin(x) dx = 0 . In general, an integral between these limits for any other
odd function will be also be zero.
In the general case we may have an integral of more than one dimension. The key to determining whether a
general integral is necessarily zero lies in the fact that because an integral is just a number, it must be invariant
to any symmetry operation. For example, bonding in a diatomic (see next section) depends on the presence of a
non-zero overlap between atomic orbitals on adjacent atoms, which may be quantified by an overlap integral. You
wouldn’t expect the bonding in a molecule to change if you rotated the molecule through some angle , so the
integral must be invariant to rotation, and indeed to any other symmetry operation. In group theoretical terms,
for an integral to be non-zero
6
, the integrand must transform as the totally symmetric irrep in the appropriate
point group. In practice, the integrand may not transform as a single irrep, but it must include the totally
symmetric irrep. These ideas should become more clear in the next section.
18. Bonding in diatomics
You will already be familiar with the idea of constructing molecular
orbitals from linear combinations of atomic orbitals from previous
courses covering bonding in diatomic molecules. By considering the
symmetries of s and p orbitals on two atoms, we can form bonding
and antibonding combinations labelled as having either or
symmetry depending on whether they resemble s or a p orbitals
when viewed along the bond axis (see diagram below). In all of the
cases shown, only atomic orbitals that have the same symmetry
when viewed along the bond
axis z can form a chemical bond e.g. two s orbitals, two p
z
orbitals ,
or an s and a p
z
can form a bond, but a p
z
and a p
x
or an s and a p
x
or
a p
y
cannot. It turns out that the rule that determines whether or
not two atomic orbitals can bond is that they must belong to the
same symmetry species within the point group of the molecule.
We can prove this mathematically for two atomic orbitals
i
and
j
by looking at the overlap integral between the two orbitals.
S
ij
= <
i
|
j
> =
i
*
j
d
In order for bonding to be possible, this integral must be non-zero.
The product of the two functions
and
2
transforms as the direct product of their symmetry species i.e.
12
=
1
2
. As explained above, for the overlap integral to be non-zero,
12
must contain the totally symmetric irrep
(A
1g
for a homonuclear diatomic, which belongs to the point group D
∞h
). As it happens, this is only possible if
1
and
2
belong to the same irrep. These ideas are summarised for a diatomic in the table below.
6
It should be noted that even when the irreps spanned by the integrand do include the totally symmetric irrep, it is still
possible for the integral to be zero. All group theory allows us to do is identify integrals that are necessarily zero based on the
symmetry (or lack thereof) of the integrand.

29
19. Bonding in polyatomics - constructing molecular orbitals from SALCs
In the previous section we showed how to use symmetry to determine whether two atomic orbitals can form a
chemical bond. How do we carry out the same procedure for a polyatomic molecule, in which many atomic orbitals
may combine to form a bond? Any SALCs of the same symmetry could potentially form a bond, so all we need to
do to construct a molecular orbital is take a linear combination of all the SALCs of the same symmetry species.
The general procedure is:
1. Use a basis set consisting of valence atomic orbitals on each atom in the system.
2. Determine which irreps are spanned by the basis set and construct the SALCs that transform as each
irrep.
3. Take linear combinations of irreps of the same symmetry species to form the molecular orbitals.
e.g. in our NH
3
example we could form a molecular orbital of A
1
symmetry from the two SALCs that
transform as A
1
,
(A
1
) = c
1
1
+ c
2
2
= c
1
s
N
+ c
2
1
3
(s
1
+s
2
+s
3
) (19.1)
Unfortunately, this is as far as group theory can take us. It can give us the functional form of the molecular
orbitals but it cannot determine the coefficients c
1
and c
2
. To go further and obtain the expansion coefficients
and orbital energies, we must turn to quantum mechanics. The material we are about to cover will be repeated in
greater detail in later courses on quantum mechanics and valence, but they are included here to provide you with a
complete reference on how to construct molecular orbitals and determine their energies.
20. Calculating the orbital energies and expansion coefficients
Note: Sections 20 and 21 are not covered in this lecture course, but the material will be dealt with in other
courses (e.g. valence) and is included here for completeness.
Calculation of the orbital energies and expansion coefficients is based on the variation principle, which states that
any approximate wavefunction must have a higher energy than the true wavefunction. This follows directly from
the fairly common-sense idea that in general any system tries to minimize its energy. If an ‘approximate’
wavefunction had a lower energy than the ‘true’ wavefunction, we would expect the system to try and adopt this
‘approximate’ lower energy state, rather than the ‘true’ state. That all approximations to the true wavefunction
must have a higher energy than the true wavefunction is the only scenario that makes physical sense. A
mathematical proof of the variation principle is given in the Appendix.
We apply the variation principle as follows:
First atomic
orbital
Second atomic
orbital
1
2
Overlap
integral
Bonding?
s (A
1g
) s (A
1g
) A
1g
Non-zero Yes
s (A
1g
)
p
x
(E
1u
)
E
1u
Zero
No
s (A
1g
) p
z
(A
1u
) A
1u
Zero No
p
x
(E
1u
)
p
x
(E
1u
)
A
1g
+ A
2g
+ E
2g
Non
-
zero
Yes
p
x
(E
1u
) p
z
(A
1u
) E
1g
Zero No
p
z
(A
1u
)
p
z
(A
1u
)
A
1g
Non
-
zero
Yes

30
Molecular energy levels, or orbital energies, are eigenvalues of the molecular Hamiltonian H
^
. Using a standard
result from quantum mechanics, it follows that the energy E of a molecular orbital is
E =
<|H
^
|>
<|>
(unnormalised ) (20.1)
or E = <|H
^
|> (normalised , for which <|> = 1) (20.2)
If the true wavefunction has the lowest energy, then to find the closest approximation we can to the true
wavefunction, all we have to do is find the coefficients in our expansion of SALCs that minimise the energy in the
above expressions. In practice, we substitute our wavefunction into Equation (19.1) and minimise the resulting
expression with respect to the coefficients. To show how this is done, we’ll use our NH
3
wavefunction of A
1
symmetry from the previous section. Substituting into Equation (20.1) gives:
E =
<c
1
1
+c
2
2
|H
^
|c
1
1
+c
2
2
>
< c
1
1
+c
2
2
| c
1
1
+c
2
2
>
=
<c
1
1
|H
^
|c
1
1
> + <c
1
1
|H
^
|c
2
2
> + <c
2
2
|H
^
|c
1
1
> + <c
2
2
|H
^
|c
2
2
>
<c
1
1
|c
1
1
> + <c
1
1
|c
2
2
> + <c
2
2
|c
1
1
> + <c
2
2
|c
2
2
>
=
c
1
2
<
1
|H
^
|
1
> + c
1
c
2
<
1
|H
^
|
2
> + c
2
c
1
<
2
|H
^
|
1
> + c
2
2
<
2
|H
^
|
2
>
c
1
2
<
1
|
1
> + c
1
c
2
<
1
|
2
> + c
2
c
1
<
2
|
1
> + c
2
2
<
2
|
2
>
If we now define a Hamiltonian matrix element H
ij
= <
i
|H
^
|
j
> and an overlap integral S
ij
= <
i
|
j
> and note that
H
ij
=H
ji
and S
ij
= S
ji
, this simplifies to
E =
c
1
2
H
11
+ 2c
1
c
2
H
12
+ c
2
2
H
22
c
1
2
S
11
+ 2c
1
c
2
S
12
+ c
2
2
S
22
To get this into a simpler form for carrying out the energy minimisation, we multiply both sides through by the
denominator to give
E (c
1
2
S
11
+ 2c
1
c
2
S
12
+ c
2
2
S
22
) = c
1
2
H
11
+ 2c
1
c
2
H
12
+ c
2
2
H
22
Now we need to minimise the energy with respect to c
1
and c
2
i.e. we require
E
c
1
= 0 and
E
c
2
= 0. If we
differentiate the above equation through separately by c
1
and c
2
and apply this condition, we will end up with two
equations in the two unknowns c
1
and c
2
, which we can solve to determine the coefficients and the energy.
Differentiating by c
1
gives
E
c
1
( c
1
2
S
11
+ 2c
1
c
2
S
12
+ c
2
2
S
22
) + E(2c
1
S
11
+ 2c
2
S
12
) = 2c
1
H
11
+ 2c
2
H
12
Differentiating by c
2
gives
E
c
2
( c
1
2
S
11
+ 2c
1
c
2
S
12
+ c
2
2
S
22
) + E(2c
1
S
12
+ 2c
2
S
22
) = 2c
1
H
12
+ 2c
2
H
22
Because
E
c
1
=
E
c
2
= 0, the first term on the left hand side of both equations is zero, leaving us with
E(2c
1
S
11
+ 2c
2
S
12
) = 2c
1
H
11
+ 2c
2
H
12
E(2c
1
S
12
+ 2c
2
S
22
) = 2c
1
H
12
+ 2c
2
H
22
These are normally rewritten slightly, in the form
31
c
1
(H
11
-ES
11
) + c
2
(H
12
-ES
12
) = 0 (20.3)
c
1
(H
12
-ES
12
) + c
2
(H
22
-ES
22
) = 0
These equations are known as the secular equations and are the set of equations we need to solve to determine c
1
,
c
2
and E. In the general case (derived in the Appendix), when our wavefunction is a linear combination of N SALCs
(i.e. =
i=1
N
c
i
i
) we get N equations in N unknowns, with the k
th
equation given by
i=1
N
c
i
(H
ki
–ES
ki
) = 0 (20.4)
Note that we can use any basis functions we like together with the linear variation method described here to
construct approximate molecular orbitals and determine their energies, but choosing to use SALCs simplifies
things considerably when the number of basis functions is large. An arbitrary set of N basis functions leads to a
set of N equations in N unknowns, which must be solved simultaneously. Converting the basis into a set of SALCs
separates the equations into several smaller sets of secular equations, one for each irrep, which can be solved
independently. It is usually easier to solve several sets of secular equations of lower dimensionality than one set
of higher dimensionality.
21. Solving the secular equations
21.1 Matrix formulation of a set of linear equations
As we have seen already, any set of linear equations may be rewritten as a matrix equation Ax = b. Linear
equations are classified as simultaneous linear equations or homogeneous linear equations, depending on whether
the vector b on the RHS of the equation is non-zero or zero.
For a set of simultaneous linear equations (non-zero b) it is fairly apparent that if a unique solution exists, it can
be found by multiplying both sides by the inverse matrix A
-1
(since A
-1
A on the left hand side is equal to the
identity matrix, which has no effect on the vector x)
Ax = b
A
-1
Ax = A
-1
b
x = A
-1
b
In practice, there are easier matrix methods for solving simultaneous equations than finding the inverse matrix,
but these need not concern us here. In Section 8.4, we discovered that in order for a matrix to have an inverse,
it must have a non-zero determinant. Since A
-1
must exist in order for a set of simultaneous linear equations to
have a solution, this means that the determinant of the matrix A must be non-zero for the equations to be
solvable.
The reverse is true for homogeneous linear equations. In this case the set of equations only has a solution if the
determinant of A is equal to zero. The secular equations we want to solve are homogeneous equations, and we will
use this property of the determinant to determine the molecular orbital energies. An important property of
homogeneous equations is that if a vector x is a solution, so is any multiple of x, meaning that the solutions (the
molecular orbitals) can be normalised without causing any problems.
21.2 Solving for the orbital energies and expansion coefficients
Recall the secular equations for the A
1
orbitals of NH
3
derived in the previous section
c
1
(H
11
-ES
11
) + c
2
(H
12
-ES
12
) = 0
c
1
(H
12
-ES
12
) + c
2
(H
22
-ES
22
) = 0

32
where c
1
and c
2
are the coefficients in the linear combination of the SALCs
1
= s
N
and
2
=
1
3
(s
1
+ s
2
+ s
3
) used to
construct the molecular orbital. Writing this set of homogeneous linear equations in matrix form gives
H
11
-ES
11
H
12
-ES
12
H
12
-ES
12
H
22
-ES
22
c
1
c
2
=
0
0
(21.2.1)
In order for the equations to have a solution, the determinant of the matrix must be equal to zero. Writing out
the determinant will give us a polynomial equation in E that we can solve to obtain the orbital energies in terms of
the Hamiltonian matrix elements H
ij
and overlap integrals S
ij
. The number of energies obtained by ‘solving the
secular determinant’ in this way is equal to the order of the matrix, in this case two.
The secular determinant for Equation (21.2.1) is (noting that S
11
= S
22
= 1 since the SALCs are normalised)
(H
11
-E)(H
22
-E) - (H
12
-ES
12
)
2
= 0
Expanding and collecting terms in E gives
E
2
(1-S
12
2
) + E(2H
12
S
12
-H
11
-H
22
) + (H
11
H
22
-H
12
2
) = 0
which can be solved using the quadratic formula to give the energies of the two molecular orbitals.
E
=
-(2H
12
S
12
-H
11
-H
22
) (2H
12
S
12
-H
11
-H
22
)
2
- 4(1-S
12
2
)(H
11
H
22
-H
12
2
)
2(1-S
12
2
)
(21.2.2)
To obtain numerical values for the energies, we need to evaluate the integrals H
11
, H
22
, H
12
, S
12
. This would be
quite a challenge to do analytically, but luckily there are a number of computer programs that can be used to
calculate the integrals. One such program gives the following values.
H
11
= -26.0000 eV
H
22
= -22.2216 eV
H
12
= -29.7670 eV
S
12
= 0.8167
When we substitute these into our equation for the energy levels, we get:
E
+
= 29.8336 eV
E
-
= -31.0063 eV
We now have the orbital energies. The next step is to find the orbital coefficients. The coefficients for an
orbital of energy E are found by substituting the energy into the secular equations and solving for the
coefficients c
i
. Since the two secular equations are not linearly independent (i.e. they are effectively only one
equation), when we solve them to find the coefficients what we will end up with is the relative values of the
coefficients. This is true in general: in a system with N coefficients, solving the secular equations will allow all N
of the coefficients c
i
to be obtained in terms of, say, c
1
. The absolute values of the coefficients are found by
normalising the wavefunction.
Since the secular equations for the orbitals of energy E
+
and E
-
are not linearly independent, we can choose to
solve either one of them to find the orbital coefficients. We will choose the first.
(H
11
- E
)c
1
+ (H
12
- E
S
12
)c
2
= 0
For the orbital with energy E
-
= -31.0063 eV, substituting numerical values into this equation gives
5.0063 c
1
– 4.4442 c
2
= 0
c
2
= 1.1265 c
1

33
The molecular orbital is therefore
1
= c
1
(
1
+1.1265
2
)
Normalising to find the constant c
1
(by requiring <|> = 1) gives
1
= 0.4933
1
+ 0.5557
2
= 0.4933 s
N
+ 0.3208 (s
1
+ s
2
+ s
3
) (substituting the SALCs for
1
and
2
)
For the second orbital, with energy E
+
= 29.8336 eV, the secular equation is
-55.8336 c
1
– 54.1321 c
2
= 0
c
2
= -1.0314 c
1
giving
2
= c
1
(
1
– 1.0314
2
)
= 1.6242
1
– 1.6752
2
(after normalisation)
= 1.6242 s
N
– 0.9672 (s
1
+ s
2
+ s
3
)
These two A
1
molecular orbitals
1
and
2
, one bonding and one antibonding, are shown below.
1
2
The remaining two SALCs arising from the s orbitals of NH
3
(
3
=
1
6
(2s
1
–s
2
–s
3
) and
4
=
1
2
(s
2
–s
3
)), form an
orthogonal pair of molecular orbitals of E symmetry. We can show this by solving the secular determinant to find
the orbital energies. The secular equations in this case are:
Solving the secular determinant gives
E
=
-(2H
34
S
34
-H
33
-H
44
) (2H
34
S
34
-H
33
-H
44
)
2
- 4(1-S
34
2
)(H
33
H
44
-H
34
2
)
2(1-S
34
2
)
The integrals required are
H
33
= -9.2892 eV
H
44
= -9.2892 eV
H
34
= 0
S
34
= 0
Using the fact that H
34
= S
34
= 0, the expression for the energies reduces to
E
=
(H
33
+H
44
) (H
33
-H
44
)
2
giving E
+
= H
33
= -9.2892 eV and E
-
= H
44
= -9.2892 eV. Each SALC therefore forms a molecular orbital by itself,
and the two orbitals have the same energy; the two SALCs form an orthogonal pair of degenerate orbitals.
These two molecular orbitals of E symmetry are shown below.
3
4

34
22. Summary of the steps involved in constructing molecular orbitals
1. Choose a basis set of functions f
i
consisting of the valence atomic orbitals on each atom in the system, or some
chosen subset of these orbitals.
2. With the help of the appropriate character table, determine which irreps are spanned by the basis set using
Equation (15.3.4) to determine the number of times a
k
that the k
th
irrep appears in the representation.
a
k
=
1
h
C
n
C
(g)
k
(g)
3. Construct the SALCs
i
that transform as each irrep using Equation (16.1)
i
=
g
k
(g) g f
i
4. Write down expressions for the molecular orbitals by taking linear combinations of all the irreps of the same
symmetry species.
5. Write down the secular equations for the system.
6. Solve the secular determinant to obtain the energies of the molecular orbitals.
7. Substitute each energy in turn back into the secular equations and solve to obtain the coefficients appearing in
your molecular orbital expressions in 4.
8. Normalise the orbitals.
23. A more complicated bonding example – the molecular orbitals of H
2
O
As another example, we will use group theory to construct the molecular orbitals of H
2
O (point group C
2v
) using a
basis set consisting of all the valence orbitals. The valence orbitals are a 1s orbital on each hydrogen, which we
will label s
H
and s
H
’, and a 2s and three 2p orbitals on the oxygen, which we will label s
O
, p
x
, p
y
, p
z
, giving a complete
basis (s
H
,s
H
’,s
O
,p
x
,p
y
,p
z
).
The first thing to do is to determine how each orbital transforms under the symmetry operations of the C
2v
point
group (E, C
2
,
v
and
v
’), construct a matrix representation and determine the characters of each operation. The
symmetry operations and axis system we will be using are shown below.
The orbitals transform in the following way
E (s
H
, s
H
’, s
O
, p
x
, p
y
, p
z
) (s
H
, s
H
’, s
O
, p
x
, p
y
, p
z
)
C
2
(s
H
, s
H
’, s
O
, p
x
, p
y
, p
z
) (s
H
‘, s
H
, s
O
, -p
x
, -p
y
, p
z
)
v
(xz) (s
H
, s
H
’, s
O
, p
x
, p
y
, p
z
) (s
H
, s
H
’, s
O
, p
x
, -p
y
, p
z
)
v
’(yz) (s
H
, s
H
’, s
O
, p
x
, p
y
, p
z
) (s
H
’, s
H
, s
O
, -p
x
, p
y
, p
z
)
A short aside on constructing matrix representatives
After a little practice, you will probably be able to write matrix representatives straight away just by looking at
the effect of the symmetry operations on the basis. However, if you are struggling a little the following
procedure might help.

35
Remember that the matrix representatives are just the matrices we would have to multiply the left hand side of
the above equations by to give the right hand side. In most cases they are very easy to work out. Probably the
most straightforward way to think about it is that each column of the matrix shows where one of the original
basis functions ends up. For example, the first column transforms the basis function s
H
to its new position. The
first column of the matrix can be found by taking the result on the right hand side of the above expressions,
replacing every function that isn’t s
H
with a zero, putting the coefficient of s
H
(1 or –1 in this example) in the
position at which it occurs, and taking the transpose to give a column vector.
e.g. Consider the representative for the C
2
operation. The original basis (s
H
, s
H
’, s
O
, p
x
, p
y
, p
z
) transforms into
(s
H
’, s
H
, s
O
, -p
x
, -p
y
, p
z
). The first column of the matrix therefore transforms s
H
into s
H
’. Taking the result
and replacing all the other functions with zeroes gives (0, s
H
, 0, 0, 0, 0). The coefficient of s
H
is 1, so the
first column of the C
2
matrix representative is
0
1
0
0
0
0
23.1 Matrix representation, characters and SALCs
The matrix representatives and their characters are
E C
2
v
v
’
1 0 0 0 0 0
0 1 0 0 0 0
0 0 1 0 0 0
0 0 0 1 0 0
0 0 0 0 1 0
0 0 0 0 0 1
0 1 0 0 0 0
1 0 0 0 0 0
0 0 1 0 0 0
0 0 0 -1 0 0
0 0 0 0 -1 0
0 0 0 0 0 1
1 0 0 0 0 0
0 1 0 0 0 0
0 0 1 0 0 0
0 0 0 1 0 0
0 0 0 0 -1 0
0 0 0 0 0 1
0 1 0 0 0 0
1 0 0 0 0 0
0 0 1 0 0 0
0 0 0 -1 0 0
0 0 0 0 1 0
0 0 0 0 0 1
(E) = 6 (C
2
) = 0 (
v
) = 4 (
v
’) = 2
Now we are ready to work out which irreps are spanned by the basis we have chosen. The character table for C
2v
is:
As before, we use Equation (15.3.4) to find out the number of times each irrep appears.
a
k
=
1
h
C
n
C
(g)
k
(g)
We have
a(A
1
) = ¼ (1x6x1 + 1x0x1 + 1x4x1 + 1x2x1) = 3
a(A
2
) = ¼ (1x6x1 + 1x0x1 + 1x4x-1 + 1x2x-1) = 0
a(B
1
) = ¼ (1x6x1 + 1x0x-1 + 1x4x1 + 1x2x-1) = 2
a(B
2
) = ¼ (1x6x1 + 1x0x-1 + 1x4x-1 + 1x2x1) = 1
so the basis spans 3A
1
+ 2B
1
+ B
2
. Now we use the projection operators applied to each basis function f
i
in turn to
determine the SALCs
i
=
g
k
(g) g f
i
The SALCs of A
1
symmetry are:
C
2v
E
C
2
v
v
’
h = 4
A
1
1 1 1 1 z, x
2
, y
2
, z
2
A
2
1
1
-
1
-
1
xy, R
z
B
1
1 -1 1 -1 x, xz, R
y
B
2
1 -1 -1 1 y, yz, R
x

36
(s
H
) = s
H
+ s
H
’ + s
H
+ s
H
’ = 2(s
H
+ s
H
’)
(s
H
’) = s
H
’ + s
H
+ s
H
’ + s
H
= 2(s
H
+ s
H
’)
(s
O
) = s
O
+ s
O
+ s
O
+ s
O
= 4s
O
(p
x
) = p
x
– p
x
+ p
x
– p
x
= 0
(p
y
) = p
y
– p
y
– p
y
+ p
y
= 0
(p
z
) = p
z
+ p
z
+ p
z
+ p
z
= 4p
z
The SALCs of B
1
symmetry are:
(s
H
) = s
H
- s
H
’ + s
H
- s
H
’ = 2(s
H
- s
H
’)
(s
H
’) = s
H
’ - s
H
+ s
H
’ - s
H
= 2(s
H
’ - s
H
)
(s
O
) = s
O
- s
O
+ s
O
- s
O
= 0
(p
x
) = p
x
+ p
x
+ p
x
+ p
x
= 4p
x
(p
y
) = p
y
+ p
y
– p
y
- p
y
= 0
(p
z
) = p
z
- p
z
+ p
z
- p
z
= 0
The SALCs of B
2
symmetry are:
(s
H
) = s
H
- s
H
’ - s
H
+ s
H
’ = 0
(s
H
’) = s
H
’ - s
H
- s
H
’ + s
H
= 0
(s
O
) = s
O
- s
O
- s
O
+ s
O
= 0
(p
x
) = p
x
+ p
x
- p
x
- p
x
= 0
(p
y
) = p
y
+ p
y
+ p
y
+ p
y
= 4p
y
(p
z
) = p
z
- p
z
- p
z
+ p
z
= 0
After normalisation, our SALCs are therefore:
A
1
symmetry
1
=
1
2
(s
H
+ s
H
’)
2
= s
O
3
= p
z
B
1
symmetry
4
=
1
2
(s
H
- s
H
’)
5
= p
x
B
2
symmetry
6
= p
y
Note that we only take one of the first two SALCs generated by the B
1
projection operator since one is a simple
multiple of the other (i.e. they are not linearly independent). We can therefore construct three molecular
orbitals of A
1
symmetry, with the general form
(A
1
) = c
1
1
+ c
2
2
+ c
3
3
= c
1
’(s
H
+ s
H
’) + c
2
s
O
+ c
3
p
z
where c
1
’ = c
1
/ 2
two molecular orbitals of B
1
symmetry, of the form
(B
1
) = c
4
4
+ c
5
5
= c
4
’(s
H
-s
H
’) + c
5
p
z
and one molecular orbital of B
2
symmetry
(B
2
) =
6

37
= p
y
To work out the coefficients c
1
-c
5
and determine the orbital energies, we would have to solve the secular
equations for each set of orbitals in turn. We are not dealing with a conjugated system, so in this case Huckel
theory cannot be used and the various H
ij
and S
ij
integrals would have to be calculated numerically and substituted
into the secular equations. This involves a lot of tedious algebra, which we will leave out for the moment. The
LCAO orbitals determined above are an approximation of the true molecular orbitals of water, which are shown on
the right. As we have shown using group theory, the A
1
molecular orbitals involve the oxygen 2s and 2p
z
atomic
orbitals and the sum s
H
+s
H
’ of the hydrogen 1s orbitals. The B
1
molecular orbitals involve the oxygen 2p
x
orbital
and the difference s
H
-s
H
’ of the two hydrogen 1s orbitals, and the B
2
molecular orbital is essentially an oxygen 2p
y
atomic orbital.
24. Molecular vibrations
Vibrational motion in diatomic molecules was introduced last year, in the context of the simple harmonic oscillator
in quantum mechanics. A diatomic molecule has only a single bond that can vibrate; we say it has a single
vibrational mode. As you may expect, the vibrational motions of polyatomic molecules are much more complicated
than those in a diatomic. Firstly, there are more bonds that can vibrate; and secondly, in addition to stretching
vibrations, the only type of vibration possible in a diatomic, we can also have bending and torsional vibrational
modes. Since changing one bond length in a polyatomic will often affect the length of nearby bonds, we cannot
consider the vibrational motion of each bond in isolation; instead we talk of normal modes involving the concerted
motion of groups of bonds. As a simple example, the normal modes of a linear triatomic molecule are shown below.
symmetr ic s tretch as ymmetric stretch bend (doubly d egenerate)
Once we know the symmetry of a molecule at its equilibrium structure, group theory allows us to predict the
vibrational motions it will undergo using exactly the same tools we used above to investigate molecular orbitals.
Each vibrational mode transforms as one of the irreps of the molecule’s point group. Before moving on to an
example, we will quickly review how to determine the number of vibrational modes in a molecule.
24.1 Molecular degrees of freedom – determining the number of normal vibrational modes
An atom can undergo only translational motion, and therefore has three degrees of freedom corresponding to
motion along the x, y, and z Cartesian axes. Translational motion in any arbitrary direction can always be
expressed in terms of components along these three axes. When atoms combine to form molecules, each atom
still has three degrees of freedom, so the molecule as a whole has 3N degrees of freedom, where N is the number
of atoms in the molecule. However, the fact that each atom in a molecule is bonded to one or more neighbouring
atoms severely hinders its translational motion, and also ties its motion to that of the atoms to which it is
attached. For these reasons, while it is entirely possible to describe molecular motions in terms of the
translational motions of individual atoms (we will come back to this in the next section), we are often more
interested in the motions of the molecule as a whole. These may be divided into three types: translational;
rotational and vibrational.
Just as for an individual atom, the molecule as a whole has three degrees of translational freedom, leaving 3N-3
degrees of freedom in rotation and vibration.
The number of rotational degrees of freedom depends on the structure of the molecule. In general, there are
three possible rotational degrees of freedom, corresponding to rotation about the x, y and z Cartesian axes. A
non-linear polyatomic molecule does indeed have three rotational degrees of freedom, leaving 3N-6 degrees of
freedom in vibration (i.e 3N-6 vibrational modes). In a linear molecule, the situation is a little different. It is
generally accepted that to be classified as a true rotation, a motion must change the position of one or more of
the atoms. If we define the z axis as the molecular axis, we see that spinning the molecule about the axis does
not move any of the atoms from their original position, so this motion is not truly a rotation. Consequently, a

38
linear molecule has only two degrees of rotational freedom, corresponding to rotations about the x and y axis.
This type of molecule has 3N-5 degrees of freedom left for vibration, or 3N-5 vibrational modes.
In summary,
A linear molecule has 3N-5 vibrational modes
A non-linear molecule has 3N-6 vibrational modes.
24.2 Determining the symmetries of molecular motions
We mentioned above that the procedure for determining the normal vibrational modes of a polyatomic molecule is
very similar to that used in previous sections to construct molecular orbitals. In fact, virtually the only
difference between these two applications of group theory is the choice of basis set.
As we have already established, the motions of a molecule may be described in terms of the motions of each atom
along the x, y and z axis. Consequently, it probably won’t come as too much of a surprise to discover that a very
useful basis for describing molecular motions comprises a set of (x, y, z) axes centred on each atom. This basis is
usually known as the 3N Cartesian basis (since there are 3N Cartesian axes, 3 axes for each of the N atoms in the
molecule). Note that each molecule will have a different 3N Cartesian basis, just as every molecule has a
different atomic orbital basis.
Our first task in investigating motions of a particular molecule is to determine the characters of the matrix
representatives for the 3N Cartesian basis under each of the symmetry operations in the molecular point group.
We will use the H
2
O molecule, which has C
2v
symmetry, as an example.
H
2
O has three atoms, so the 3N Cartesian basis will have 9 elements. The basis vectors are shown in the diagram
below.
x
H
y
H
x
H’
y
H ’
z
H ’
x
O
y
O
z
O
z
H
One way of determining the characters would be to construct all of the matrix representatives and take their
traces. While you are more than welcome to try this approach if you want some practice at constructing matrix
representatives, there is an easier way. Recall that we can also determine the character of a matrix
representative under a particular symmetry operation by stepping through the basis functions and applying the
following rules:
i) Add 1 to the character if the basis function is unchanged by the symmetry operation;
ii) Add –1 to the character if the basis function changes sign under the symmetry operation;
iii) Add 0 to the character if the basis function moves when the symmetry operation is applied.
For H
2
O, this gives us the following characters for the 3N Cartesian basis (check that you can obtain this result
using the rules above and the basis vectors as drawn in the figure):
Operation: E C
2
v
(xz)
v
’(yz)
3N
: 9 -1 3 1
There is an even quicker way to work out the characters of the 3N Cartesian basis if you have a character table in
front of you. The character for the Cartesian basis is simply the sum of the characters for the x, y and z (or T
x
,
T
y
, and T
z
) functions listed in the character table. To get the character for the 3N Cartesian basis, simply
multiply this by the number of atoms in the molecule that are unshifted by the symmetry operation.
The C
2v
character table is shown below.

39
x transforms as B
1
, y as B
2
, and z as A
1
, so the characters for the Cartesian basis are
Operation: E C
2
v
(xz)
v
’(yz)
Cart
: 3 -1 1 1
We multiply each of these by the number of unshifted atoms (3 for the identity operation, 1 for C
2
, 3 for
v
and 1
for
v
’) to obtain the characters for the 3N Cartesian basis.
3N
: 9 -1 3 1
Reassuringly, we obtain the same characters as we did previously. Which of the three methods you use to get to
this point is up to you.
We now have the characters for the molecular motions (described by the 3N Cartesian basis) under each
symmetry operation. At this point, we want to separate these characters into contributions from translation,
rotation, and vibration. This turns out to be a very straightforward task. We can read the characters for the
translational and rotational modes directly from the character table, and we obtain the characters for the
vibrations simply by subtracting these from the 3N Cartesian characters we’ve just determined. The characters
for the translations are the same as those for
Cart
. We find the characters for the rotations by adding together
the characters for R
x
, R
y
and R
z
from the character table (or just R
x
and R
y
if the molecule is linear). For H
2
O, we
have:
Operation: E C
2
v
(xz)
v
’(yz)
3N
: 9 -1 3 1
Trans
: 3 -1 1 1
Rot
: 3 -1 -1 -1
Vib
=
3N
-
Trans
-
Rot
: 3 1 3 1
The characters in the final row are the sums of the characters for all of the molecular vibrations. We can find
out the symmetries of the individual vibrations by using the reduction equation (equation 15.3.4) to determine the
contribution from each irrep.
In many cases you won’t even need to use the equation, and can work out which irreps are contributing just by
inspection of the character table. In the present case, the only combination of irreps that can give the required
values for
Vib
is 2A
1
+ B
1
. As an exercise, you should make sure you are also able to obtain this result using the
reduction equation.
So far this may all seem a little abstract, and you probably want to know is what the vibrations of H
2
O actually
look like. For a molecule with only three atoms, it is fairly easy to identify the possible vibrational modes and to
assign them to the appropriate irrep.
A
1
B
1
A
1
symm etr ic stretch asym metric stretch bend
For a larger molecule, the problem may become much more complex, and in that case we can generate the SALCs
of the 3N Cartesian basis, which will tell us the atomic displacements associated with each vibrational mode. We
will do this now for H
2
O.
C
2v
E
C
2
v
v
’
h = 4
A
1
1
1
1
1
z, x
2
, y
2
, z
2
A
2
1 1 -1 -1 xy, R
z
B
1
1
-
1
1
-
1
x, xz, R
y
B
2
1 -1 -1 1 y, yz, R
x
40
24.3 Atomic displacements using the 3N Cartesian basis
As before, we generate the SALCs of each symmetry by applying the appropriate projection operator to each of
the basis functions (or in this case, basis vectors) f
i
in turn.
i
=
g
k
(g) g f
i
In this case we have 9 basis vectors, which we will label x
H
, y
H
, z
H
, x
O
, y
O
, z
O
, x
H’
, y
H’
, z
H’
, describing the
displacements of the two H atoms and the O atom along Cartesian axes. For the SALCs of A
1
symmetry, applying
the projection operator to each basis vector in turn gives (check that you can obtain this result):
1
(x
H
) = x
H
– x
H’
+ x
H
– x
H’
=2x
H
– 2x
H’
2
(y
H
) = y
H
– y
H’
– y
H
+ y
H’
= 0
3
(z
H
) = z
H
+ z
H’
+ z
H
+ z
H’
= 2z
H
+ 2z
H’
4
(x
O
) = x
O
– x
O
+ x
O
– x
O
= 0
5
(y
O
) = y
O
– y
O
– y
O
+ y
O
= 0
6
(z
O
) = z
O
+ z
O
+ z
O
+ z
O
= 4z
O
7
(x
H’
) = x
H’
– x
H
+ x
H’
– x
H
=2x
H’
– 2x
H
8
(y
H’
) = y
H’
– y
H
– y
H’
+ y
H
= 0
9
(z
H’
) = z
H’
+ z
H
+ z
H’
+ z
H
= 2z
H’
+ 2z
H
We see that the motion characteristic of an A
1
vibration (which we have identified as the symmetric stretch and
the bending vibration) may be summarised as follows:
i) 2(x
H
-x
H’
) - the two hydrogen atoms move in opposite directions along the x axis.
ii) 2(z
H
+z
H’
) – the two hydrogen atoms move in the same direction along the z axis.
iii) 4z
O
- the oxygen atom moves along the z axis.
iv) There is no motion of any of the atoms in the y direction.
The asymmetric stretch has B
1
symmetry, and applying the projection operator in this case gives:
1
(x
H
) = x
H
+ x
H’
+
xH
+ x
H’
=2x
H
+ 2x
H’
2
(y
H
) = y
H
+ y
H’
– y
H
- y
H’
= 0
3
(z
H
) = z
H
- z
H’
+ z
H
- z
H’
= 2z
H
– 2z
H’
4
(x
O
) = x
O
+ x
O
+ x
O
+ x
O
= 4x
O
5
(y
O
) = y
O
+ y
O
– y
O
- y
O
= 0
6
(z
O
) = z
O
- z
O
+ z
O
- z
O
= 0
7
(x
H’
) = x
H’
+ x
H
+ x
H’
+ x
H
=2x
H’
+ 2x
H
8
(y
H’
) = y
H’
+ y
H
– y
H’
- y
H
= 0
9
(z
H’
) = z
H’
- z
H
+ z
H’
- z
H
= 2z
H’
– 2z
H
In this vibrational mode, the two H atoms move in the same direction along the x axis and in opposite directions
along the z axis.
We have now shown how group theory may be used together with the 3N Cartesian basis to identify the
symmetries of the translational, rotational and vibrational modes of motion of a molecule, and also to determine
the atomic displacements associated with each vibrational mode.
24.4 Molecular vibrations using internal coordinates
While it was fairly straightforward to investigate the atomic displacements associated with each vibrational mode
of H
2
O using the 3N Cartesian basis, this procedure becomes more complicated for larger molecules. Also, we
are often more interested in how bond lengths and angles change in a vibration, rather than in the Cartesian
displacements of the individual atoms. If we are only interested in looking at molecular vibrations, we can use a
different procedure from that described above, and start from a basis of internal coordinates. Internal
coordinates are simply a set of bond lengths and bond angles, which we can use as a basis for generating

41
representations and, eventually, SALCs. Since bond lengths and angles do not change during translational or
rotational motion, no information will be obtained on these types of motion.
For H
2
O, the three internal coordinates of interest are the two OH bond lengths, which we
will label r and r’, and the HOH bond angle, which we will label . If we wanted to, we could
separate our basis into two different bases, one consisting only of bond lengths, to
describe stretching vibrations, and one consisting of only bond angles, to describe bending
vibrations. However, the current example is simple enough to treat all the basis functions together.
As usual, our first step is to work out the characters of the matrix representatives for this basis under each
symmetry operation. The effects of the various transformations on our chosen basis, and the characters of the
corresponding representatives, are:
E(r,r’,) = (r,r’,) (E) = 3
C
2
(r,r’,) = (r’,r,) (C
2
) = 1
v
(xz)(r,r’,) = (r,r’,) (
v
) = 3
v
’(yz)(r,r’,) = (r’,r,) (
v
’) = 1
These are the same characters as we found before using the 3N Cartesian basis, and as before, we can see by
inspection of the character table that the representation may be reduced down to the sum of irreps 2A
1
+ B
1
. We
can now work out the symmetry adapted linear combinations of our new basis set to see how the bond lengths and
angle change as H
2
O vibrates in each of the three vibrational modes.
Again, we will use the projection operator
i
=
g
k
(g) g f
i
applied to each basis function in turn.
Firstly the A
1
vibrations:
1
(r) = r + r’ + r + r’ = 2(r + r’)
2
(r’) = r’ + r +r’ + r = 2(r + r’)
3
() = + + + = 4
From these SALCs, we can identify
1
(and
2
, which is identical) with the symmetric stretch, in which both bond
lengths change in phase with each other, and
3
with the bend.
Now for the B
1
vibration:
(r) = r – r’ + r – r’ = 2(r – r’)
5
(r’) = r’ – r + r’ – r = 2(r’ – r)
6
() = – + – = 0
4
and
5
are not linearly independent, and either one may be chosen to describe the asymmetric stretch, in which
one bond lengthens as the other shortens.
Note: When using internal coordinates, it is important that all of the coordinates in the basis are linearly
independent. If this is the case then the number of internal coordinates in the basis will be the same as the
number of vibrational modes (3N-5 or 3N-6, depending on whether the molecule is linear or non-linear). This
requirement is satisfied in the H
2
O example above. For a less straightforward example, consider the methane
molecule, CH
4
. It might appear that we could choose a basis made up of the four C-H bond lengths and the six H-
C-H bond angles. However, this would give us 10 basis functions, and CH
4
has only 9 vibrational modes. This is due
to the fact that the bond angles are not all independent of each other. It can be tricky to come up with the
appropriate internal coordinate basis to describe all of the molecular motions, but all is not lost. Even if you can’t
work out the appropriate bond angles to choose, you can always take a basis of bond lengths to investigate the
stretching vibrations of a molecule. If you want to know the symmetries of the bending vibrations, you can use
the 3N Cartesian basis method to determine the symmetries of all of the vibrational modes and compare these
with the stretching mode symmetries to identify the bending modes.
r
1
r
2
42
25. Summary of applying group theory to molecular motions
1. Atomic or molecular translations transform in the same way as the x, y, z (or T
x
, T
y
, T
z
) functions listed in
the character tables.
2. Molecular rotations transform in the same way as the R
x
, R
y
, R
z
functions listed in the character tables.
3. The irreps spanned by the motions of a polyatomic molecule may be determined using the 3N Cartesian
basis, made up of x,y,z axes on each atom. The characters of the matrix representatives are best
determined using a table as follows:
Operation: List the symmetry operations in the point group
Cart
List the characters for x + y + z (from the character table) for each operation
N
unshifted
List the number of atoms in the molecule that are unshifted by each symmetry operation
3N
Take the product of the previous two rows to give the characters for
3N
.
4. The irreps spanned by the molecular vibrations are determined by first subtracting the characters for
rotations and translations from the characters for
3N
to give the characters for
vib
and then using the
reduction formula or inspection of the character table to identify the irreps contributing to
vib
.
5. The molecular displacements for the vibrations of each symmetry may be determined by using projection
operators on the 3N Cartesian basis vectors to generate SALCs.
6. Alternatively, a basis of internal coordinates (bond lengths and angles) may be used to investigate
stretching and bending vibrations. Determine the characters, identify the irreps, and construct SALCs.
26. Group theory and molecular electronic states
Firstly, it is important that you understand the difference between a molecular orbital and an electronic state.
A strict definition of a molecular orbital is that it is a ‘one electron wavefunction’, i.e. a solution to the
Schrodinger equation for the molecule. A complete one electron wavefunction (orbital) is a product of a spatial
function, describing the orbital angular momentum and ‘shape’ of the orbital, and a spin function, describing the
spin angular momentum.
=
spatial
spin
In common usage, the word ‘orbital’ is often used to refer only to the spatial part of the ‘true’ orbital. For
example, in atoms we generally talk about ‘s orbitals’ or ‘p orbitals’ rather than ‘s spatial wavefunctions’ and ‘p
spatial wavefunctions’. In this context, two electrons with opposite spins may occupy one spatial orbital. A more
rigorous way of saying this would be to state that a given spatial wavefunction may be paired with two different
spin wavefunctions (one corresponding to a ‘spin up’ electron and one to a ‘spin down’ electron).
An electronic state is defined by the electron configuration of the system, and by the quantum numbers of each
electron contributing to that configuration. Each electronic state corresponds to one of the energy levels of the
molecule. These energy levels will obviously depend on the molecular orbitals that are occupied, and their
energies, but they also depend on the way in which the electrons within the various molecular orbitals interact
with each other. Interactions between the electrons are essentially determined by the relative orientations of
the magnetic moments associated with their orbital and spin angular momenta, which is where the dependence on
quantum numbers comes in. A given electron configuration will often give rise to a number of different electronic
states if the electrons may be arranged in different ways (with different quantum numbers) within the occupied
orbitals.
Last year you were introduced to the idea of atomic states, and learnt how to label the states arising from a given
electron configuration using term symbols of the form
2S+1
L
J
. Term symbols of this form define the spin, orbital
and total angular momenta of the state, which in turn determine its energy. Molecular states, containing

43
contributions from a number of molecular orbitals, are more complicated. For example, a given molecular orbital
will generally contain contributions from several different atomic orbitals, and as a result, electrons cannot easily
be assigned an l quantum number. Instead of using term symbols, molecular states are usually labelled according
to their symmetry (the exception to this is linear molecules, for which conventional term symbols may still be
used, albeit with a few modifications from the atomic case).
We can determine the symmetry of an electronic state by taking the direct product of the irreps for all of the
electrons involved in that state (the irrep for each electron is simply the irrep for the molecular orbital that it
occupies). Usually we need only consider unpaired electrons. Closed shell species, in which all electrons are
paired, almost always belong to the totally symmetric irrep in the point group of the molecule.
As an example, the figure on the left shows the molecular
orbitals of butadiene, which belongs to the C
2h
point group.
Since all electrons are paired, the overall symmetry of the state
is A
g
, and the label for the state once the spin multiplicity is
included is
1
A
g
. We could have arrived at the same result by
taking the direct product of the irreps for each electron. There
are two electrons in orbitals with A
u
symmetry, and two in
orbitals with B
g
symmetry, so overall we have:
A
u
A
u
B
g
B
g
= A
g
27. Spectroscopy – interaction of atoms and molecules with light
In our final application of group theory, we will investigate the way in which symmetry considerations influence
the interaction of light with matter. We have already used group theory to learn about the molecular orbitals in
a molecule. In this section we will show that it may also be used to predict which electronic states may be
accessed by absorption of a photon. We may also use group theory to investigate how light may be used to excite
the various vibrational modes of a polyatomic molecule.
Last year, you were introduced to spectroscopy in the context of electronic transitions in atoms. You learnt that
a photon of the appropriate energy is able to excite an electronic transition in an atom, subject to the following
selection rules:
n = integer
l = ±1
L = 0, ±1
S = 0
J = 0, ±1; J=0 <-->x J=0
What you may not have learnt is where these selection rules come from. In general, different types of
spectroscopic transition obey different selection rules. The transitions you have come across so far involve
changing the electronic state of an atom, and involve absorption of a photon in the UV or visible part of the
electromagnetic spectrum. There are analogous electronic transitions in molecules, which we will consider in more
detail shortly. Absorption of a photon in the infrared (IR) region of the spectrum leads to vibrational excitation
in molecules, while photons in the microwave (MW) region produce rotational excitation. Each type of excitation
obeys its own selection rules, but the general procedure for determining the selection rules is the same in all
cases. It is simply to determine the conditions under which the probability of a transition is not identically zero.
The first step in understanding the origins of selection rules must therefore be to learn how transition
probabilities are calculated. This requires some quantum mechanics.
Last year, you learnt about operators, eigenvalues and eigenfunctions in quantum mechanics. You know that if a
function is an eigenfunction of a particular operator, then operating on the eigenfunction with the operator will
return the observable associated with that state, known as the eigenvalue (i.e. A
^
= a. What you may not know
is that operating on a function that is NOT an eigenfunction of the operator leads to a change in state of the
system. In the transitions we will be considering, the molecule interacts with the electric field of the light (as
E
n
e
r
g
y
2 g
(B )
2 u
(A )
1 u
(A )
1 g
(B )

44
opposed to NMR spectroscopy, in which the nuclei interact with the magnetic field of the electromagnetic
radiation). These transitions are called electric dipole transitions, and the operator we are interested in is the
electric dipole operator, usually given the symbol
^
., which describes the electric field of the light.
If we start in some initial state
i
, operating on this state with
^
gives a new state, =
^
i
. If we want to know
the probability of ending up in some particular final state
f
, the probability amplitude is simply given by the
overlap integral between and
f
. This probability amplitude is called the transition dipole moment, and is given
the symbol
fi.
.
^
fi
= <
f
|> = <
f
|
^
|
i
>
Physically, the transition dipole moment may be thought of as describing the ‘kick’ the electron receives or
imparts to the electric field of the light as it undergoes a transition. The transition probability is given by the
square of the probability amplitude.
P
fi
=
^
fi
2
= |<
f
|
^
|
i
>|
2
Hopefully it is clear that in order to determine the selection rules for an electric dipole transition between states
i
and
f
, we need to find the conditions under which
fi
can be non-zero. One way of doing this would be to write
out the equations for the two wavefunctions (which are functions of the quantum numbers that define the two
states) and the electric dipole moment operator, and just churn through the integrals. By examining the result, it
would then be possible to decide what restrictions must be imposed on the quantum numbers of the initial and
final states in order for a transition to be allowed, leading to selection rules of the type listed above for atoms.
However, many selection rules may be derived with a lot less work, based simply on symmetry considerations.
In section 17, we showed how to use group theory to determine whether or not an integral may be non-zero. This
forms the basis of our consideration of selection rules.
27.1 Electronic transitions in molecules
Assume that we have a molecule in some initial state
i
. We want to determine which final states
f
can be
accessed by absorption of a photon.
Recall that for an integral to be non-zero, the representation for the integrand must contain the totally
symmetric irrep. The integral we want to evaluate is
^
fi
= ∫
f
*
^
i
d
so we need to determine the symmetry of the function
f
*
^
i
. As we learnt in Section 18, the product of two
functions transforms as the direct product of their symmetry species, so all we need to do to see if a transition
between two chosen states is allowed is work out the symmetry species of
f
,
^
and
i
, take their direct product,
and see if it contains the totally symmetric irrep for the point group of interest. Equivalently (as explained in
Section 18), we can take the direct product of the irreps for
^
and
i
and see if it contains the irrep for
f
. This
is best illustrated using a couple of examples.
Earlier in the course, we learnt how to determine the symmetry molecular orbitals. The symmetry of an electronic
state is found by identifying any unpaired electrons and taking the direct product of the irreps of the molecular
orbitals in which they are located. The ground state of a closed-shell molecule, in which all electrons are paired,
always belongs to the totally symmetric irrep
7
. As an example, the electronic ground state of NH
3
, which belongs
to the C
3v
point group, has A
1
symmetry. To find out which electronic states may be accessed by absorption of a
photon, we need to determine the irreps for the electric dipole operator
^
. Light that is linearly polarised along
the x, y, and z axes transforms in the same way as the functions x, y and z in the character table
8
. From the C
3v
7
It is important not to confuse molecular orbitals (the energy levels that individual electrons may occupy within the molecule)
with electronic states (arising from the different possible arrangements of all the molecular electrons amongst the molecular
orbitals). e.g. the electronic states of NH
3
are NOT the same thing as the molecular orbitals we derived earlier in the course.
These orbitals were an incomplete set, based only on the valence s electrons in the molecule. Inclusion of the p electrons is
required for a full treatment of the electronic states. The H
2
O example above should hopefully clarify this point.
8
‘x-polarised’ means that the electric vector of the light (an electromagnetic wave) oscillates along the direction of the x axis.

45
character table, we see that x- and y-polarised light transforms as E, while z-polarised light transforms as A
1
.
Therefore:
i) For x- or y-polarised light,
^
1
transforms as E A
1
= E. This means that absorption of x- or y-
polarised light by ground-state NH
3
(see figure below left) will excite the molecule to a state of E
symmetry.
ii) For z-polarised light,
^
1
transforms as A
1
A
1
= A
1
. Absorption of z-polarised light by ground
state NH
3
(see figure below right) will excite the molecule to a state of A
1
symmetry.
x
y
z
x, y polarised light z polarised light
Of course, the photons must also have the appropriate energy, in addition to having the correct polarisation to
induce a transition.
We can carry out the same analysis for H
2
O, which belongs to the C
2v
point group. We showed previously that
H
2
O has three molecular orbitals of A
1
symmetry, two of B
1
symmetry, and one of B
2
symmetry, with the ground
state having A
1
symmetry. In the C
2v
point group, x-polarised light has B
1
symmetry, and can therefore be used to
excite electronic states of this symmetry; y-polarised light has B
2
symmetry, and may be used to access the B
2
excited state; and z-polarised light has A
1
symmetry, and may be used to access higher lying A
1
states. Consider
our previous molecular orbital diagram for H
2
O.
b o nding
a nt ib onding
2B
1
3A
1
1A
1
1B
2
2A
1
1B
1
H O
2
The electronic ground state has two electrons in a B
2
orbital, giving a state of A
1
symmetry (B
2
B
2
= A
1
). The
first excited electronic state has the configuration (1B
2
)
1
(3A
1
)
1
and its symmetry is B
2
A
1
= B
2
. It may be
accessed from the ground state by a y-polarised photon (see left). The second
excited state is accessed from the ground state by exciting an electron to the 2B
1
orbital. It has the configuration (1B
2
)
1
(2B
1
)
1
, its symmetry is B
2
B
1
= A
2
. Since
neither x-, y- or z-polarised light transforms as A
2
, this state may not be excited
from the ground state by absorption of a single photon.
27.2 Vibrational transitions in molecules
Similar considerations apply for vibrational transitions. Light polarised along the x, y and z axes of the molecule
may be used to excite vibrations with the same symmetry as the x, y and z functions listed in the character table.
For example, in the C
2v
point group, x-polarised light may be used to excite vibrations of B
1
symmetry, y-polarised
light to excite vibrations of B
2
symmetry, and z-polarised light to excite vibrations of A
1
symmetry. In H
2
O, we
would use z-polarised light to excite the symmetric stretch and bending modes, and x-polarised light to excite the
asymmetric stretch. Shining y-polarised light onto a molecule of H
2
O would not excite any vibrational motion.
y polarised light
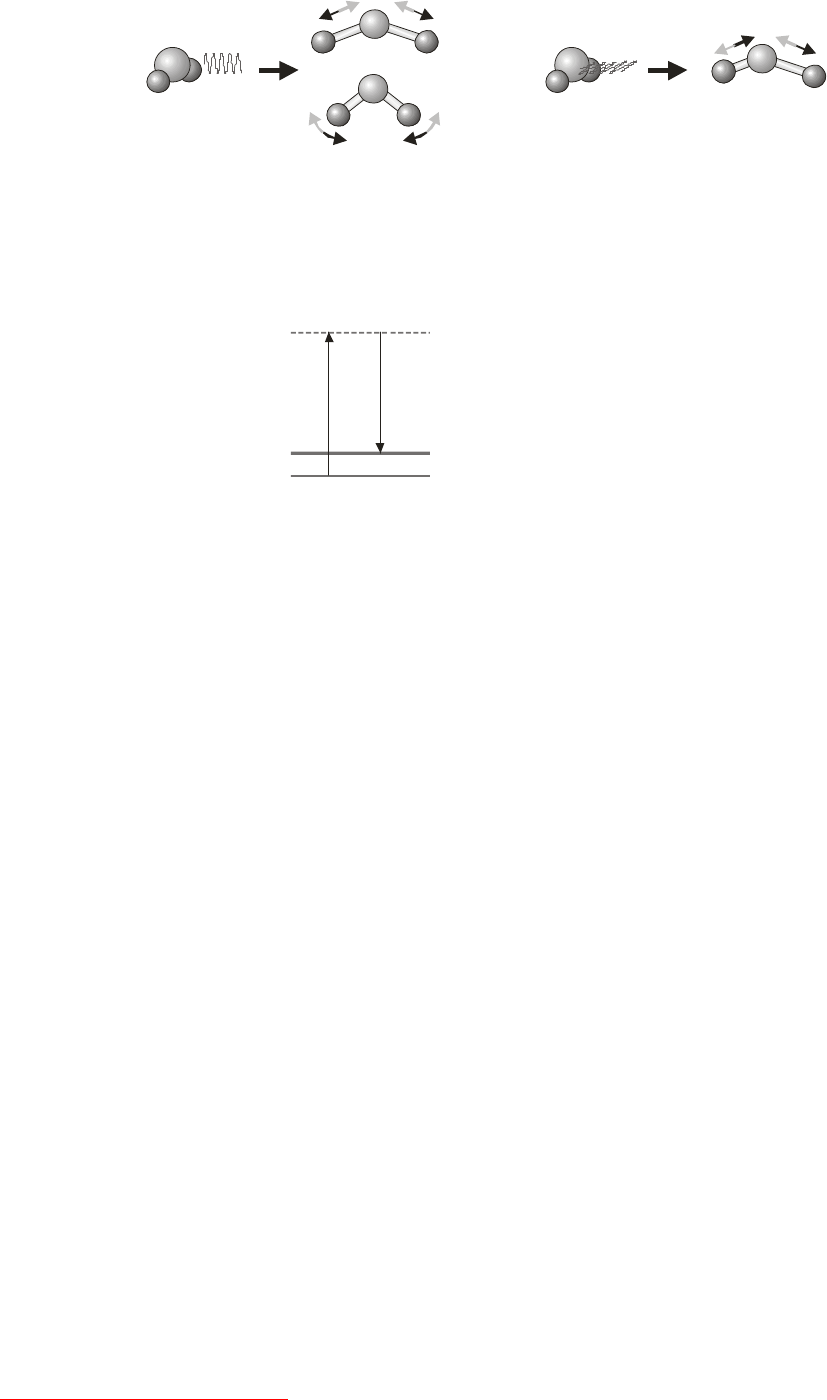
46
B
1
A
1
27.3 Raman scattering
If there are vibrational modes in the molecule that may not be accessed using a single photon, it may still be
possible to excite them using a two-photon process known as Raman scattering
9
. An energy level diagram for
Raman scattering is shown below.
initi al st at e
fin al st at e
vi rtua l s ta te
ab sor bed
photon
emitted
photon
The first photon excites the molecule to some high-lying intermediate state, known as a virtual state. Virtual
states are not true stationary states of the molecule (i.e. they are not eigenfunctions of the molecular
Hamiltonian), but they can be thought of as stationary states of the ‘photon + molecule’ system. These types of
states are extremely short lived, and will quickly emit a photon to return the system to a stable molecular state,
which may be different from the original state. Since there are two photons (one absorbed and one emitted)
involved in Raman scattering, which may have different polarisations, the transition dipole for a Raman transition
transforms as one of the Cartesian products x
2
, y
2
, z
2
, xy, xz, yz listed in the character tables. Vibrational modes
that transform as one of the Cartesian products may be excited by a Raman transition, in much the same way as
modes that transform as x, y or z may be excited by a one-photon vibrational transition.
In H
2
O, all of the vibrational modes are accessible by ordinary one-photon vibrational transitions. However, they
may also be accessed by Raman transitions. The Cartesian products transform as follows in the C
2v
point group.
A
1
x
2
, y
2
, z
2
B
1
xz
A
2
xy B
2
yz
The symmetric stretch and the bending vibration of water, both of A
1
symmetry, may therefore be excited by any
Raman scattering process involving two photons of the same polarisation (x-, y- or z-polarised). The asymmetric
stretch, which has B
1
symmetry, may be excited in a Raman process in which one photon is x-polarised and the
other z-polarised.
28. Summary
Hopefully this course has given you a reasonable introduction to the qualitative description of molecular
symmetry, and also to the way in which it can be used quantitatively within the context of group theory to predict
important molecular properties.
These main things you should have learnt in this course are:
1. How to identify the symmetry elements possessed by a molecule and assign it to a point group.
2. The consequences of symmetry for chirality and polarity of molecules.
9
You will cover Raman scattering (also known as Raman spectroscopy) in more detail in later courses. The aim here is really just
to alert you to its existence and to show how it may be used to access otherwise inaccessible vibrational modes.
47
3. The effect of applying two or more symmetry operations consecutively (group multiplication)
4. How to construct a matrix representation of a group, starting from a suitable set of basis functions.
5. How to determine the irreducible representations (irreps) spanned by a basis set, and construct symmetry
adapted linear combinations (SALCs) of the original basis functions that transform as the irreps of the group.
6. How to construct molecular orbitals by taking linear combinations of SALCs of the same symmetry species.
(7. How to set up and solve the secular equations for the molecule in order to find the molecular energy levels and
orbital coefficients – “Extra for experts”, though you will cover this in later courses)
8. How to determine the symmetries of the various modes of motion (translational, rotational and vibrational) of a
polyatomic molecule, and the symmetries of individual vibrational modes.
9. How to determine the atomic displacements in a given vibrational mode by constructing SALCs in the 3N
Cartesian basis.
10. How to determine atomic displacements in stretching and bending vibrations using internal coordinates.
11. The consequences of symmetry for the selection rules governing excitation to different electronic and
vibrational states.

48
29. Appendix A – a few proofs for the mathematically inclined
1. Proof that the character of a matrix representative is invariant under a similarity transform.
A property of traces of matrix products is that they are invariant under cyclic permutation of the matrices.
i.e. tr[ABC] = tr[BCA] = tr[CAB]. For the character of a matrix representative of a symmetry operation g, we
therefore have:
(g) = tr[(g)] = tr[C’(g)C
-1
] = tr[’(g)C
-1
C] = tr[’(g)] = ’(g)
The trace of the similarity transformed representative is therefore the same as the trace of the original
representative.
2. Proof that the characters of two symmetry operations in the same class are identical
The formal requirement for two symmetry operations g and g’ to be in the same class is that there must be some
symmetry operation f of the group such that g’=f
-1
gf (the elements g and g’ are then said to be conjugate). If we
consider the characters of g and g’ we find:
(g’) = tr[(g’)] = tr[
-1
(f)(g)(f)] = tr[(g)(f)
-1
(f)] = tr[(g)] = (g)
The characters of g and g’ are identical.
3. Proof of the variation theorem.
The variation theorem states that given a system with a Hamiltonian H, then if is any normalised, well-behaved
function that satisfies the boundary conditions of the Hamiltonian, then
<|H|> E
o
(1)
where E
0
is the true value of the lowest energy eigenvalue of H. This principle allows us to calculate an upper
bound for the ground state energy by finding the trial wavefunction for which the integral is minimised (hence
the name; trial wavefunctions are varied until the optimum solution is found). Let us first verify that the
variational principle is indeed correct.
We first define an integral
I = <|-E
0
|>
= <|H|> - <|E
0
|>
= <|H|> - E
0
<|>
= <|H|> - E
0
(since is normalised)
If we can prove that I 0 then we have proved the variation theorem.
Let
i
and E
i
be the true eigenfunctions and eigenvalues of H, so H
i
= E
i
i
. Since the eigenfunctions
i
form a
complete basis set for the space spanned by H, we can expand any wavefunction in terms of the
i
(so long as
satisfies the same boundary conditions as
i
).
=
k
a
k
k
Substituting this function into our integral I gives
I =
k
a
k
k
| H-E
0
|
j
a
j
j

49
=
k
a
k
k
|
j
(H-E
0
)
a
j
j
If we now use H = E, we obtain
I =
k
a
k
k
|
j
a
j
(E
j
-E
0
)
j
=
k
j
a
k
*a
j
(E
j
-E
0
)
k
|
j
=
k
j
a
k
*a
j
(E
j
-E
0
)
jk
We now perform the sum over j, losing all terms except the j=k term, to give
I =
k
a
k
*a
k
(E
k
-E
0
)
=
k
a
k
|
2
(E
k
-E
0
)
Since E
0
is the lowest eigenvalue, E
k
-E
0
must be positive, as must |a
k
|
2
. This means that all terms in the sum are
non-negative and I 0 as required.
For wavefunctions that are not normalised, the variational integral becomes:
|H|
|
E
0
4. Derivation of the secular equations – the general case of the linear variation method
In the study of molecules, the variation principle is often used to determine the coefficients in a linear variation
function, a linear combination of n linearly independent functions f
1
, f
2
, ..., f
n
(often atomic orbitals) that satisfy
the boundary conditions of the problem. i.e. =
i
c
i
f
i
. The coefficients c
i
are parameters to be determined by
minimising the variational integral. In this case, we have:
|H| =
i
c
i
f
i
|H|
j
c
j
f
j
=
i
j
c
i
*c
j
f
i
|H|f
j
=
i
j
c
i
*c
j
H
ij
where H
ij
is the Hamiltonian matrix element.
| =
i
c
i
f
i
|
j
c
j
f
j
=
i
j
c
i
*c
j
f
i
|f
j
=
i
j
c
i
*c
j
S
ij
where S
ij
is the overlap matrix element.
The variational energy is therefore
E =
i
j
c
i
*c
j
H
ij
i
j
c
i
*c
j
S
ij
which rearranges to give
E
i
j
c
i
*c
j
S
ij
=
i
j
c
i
*c
j
H
ij
We want to minimise the energy with respect to the linear coeffients c
i
, requiring that
E
c
i
= 0 for all i.
Differentiating both sides of the above expression gives,
E
c
k
i
j
c
i
*c
j
S
ij
+ E
i
j
c
i
*
c
k
c
j
+
c
j
c
k
c
i
* S
ij
=
i
j
c
i
*
c
k
c
j
+
c
j
c
k
c
i
* H
ij
Since
c
i
*
c
k
=
ik
and S
ij
= S
ji
, H
ij
=H
ji
, we have

50
E
c
k
i
j
c
i
*c
j
S
ij
+ 2E
i
S
ik
= 2
i
c
i
H
ik
When
E
c
k
= 0, this gives
i
c
i
(H
i k
-ES
ik
) = 0 for all k SECULAR EQUATIONS

51
30. Appendix B – Character tables and direct products
1. Character tables from from http://wulfenite.fandm.edu/Data%20/Data.html
Non axial groups
C
n
groups
C
nv
groups

52
C
nh
groups
D
n
groups
D
nh
groups

53
D
nd
groups
C
v
and D
h
S
n
groups

54
Cubic groups
55
2. Direct product tables
For the point groups O and T
d
(and O
h
)
A
1
A
2
E T
1
T
2
A
1
A
1
A
2
E T
1
T
2
A
2
A
1
E T
2
T
1
E A
1
+A
2
+E T
1
+T
2
T
1
+T
2
T
1
A
1
+E+T
1
+T
2
A
2
+E+T
1
+T
2
T
2
A
1
+E+T
1
+T
2
For the point groups D
4
, C
4v
, D
2d
(and D
4h
= D
4
C
i
)
A
1
A
2
B
2
B
2
E
A
1
A
1
A
2
B
1
B
2
E
A
2
A
1
B
2
B
1
E
B
1
A
1
A
2
E
B
2
A
1
E
E A
1
+A
2
+B
1
+B
2
For the point groups D
3
and C
3v
A
1
A
2
E
A
1
A
1
A
2
E
A
2
A
1
E
E A
1
+A
2
+E
For the point groups D
6
, C
6v
and D
3h
*
A
1
A
2
B
1
B
2
E
1
E
2
A
1
A
1
A
2
B
1
B
2
E
1
E
2
A
2
A
1
B
2
B
1
E
1
E
2
B
1
A
1
A
2
E
2
E
1
B
2
A
1
E
2
E
1
E
|
A
1
+A
2
+E
2
B
1
+B
2
+E
1
E
2
A
1
+A
2
+E
2
*in D3h make the following changes in the above table
In table In D
3h
A
1
A
1
’
A
2
A
2
’
B
1
A
1
’’
B
2
A
2
’’
E
1
E’’
E
2
E’
56
PROBLEM SHEET – MOLECULAR SYMMETRY, GROUP THEORY, & APPLICATIONS
Q1. Draw sketches to illustrate the following symmetry elements:
a) a vertical mirror plane and a C
2
axis in O
3
(ozone)
b) a horizontal mirror plane in CO
2
c) an S
4
axis in methane
d) all of the symmetry elements in CH
3
F (point group C
3v
)
e) all of the symmetry elements in ethene (point group D
2h
)
Q2. Determine the symmetry elements possessed by an s orbital, a p orbital, a d
z
2
orbital, and a d
xy
orbital
Q3. Which of the following molecules has i) a centre of inversion and ii) an S
4
axis?
a) CO
2
b) C
2
H
2
c) BF
3
d) SO
4
2-
Q4. Identify the symmetry elements in the following molecules, and assign each one to a point group (use the
flow diagram in the lecture notes if you find this helpful).
a) NH
2
Cl
b) SiF
4
c) H-CN
d) SiFClBrI
e) NO
2
f) H
2
O
2
Q5. a) What are the symmetry elements that prevent a molecule from being polar? Which of the molecules
in Q4 are polar?
b) What are the symmetry elements that exclude chirality? Which (if any) of the molecules in Q4 may
be chiral?
Q6. What are the symmetry operations in the point group C
2v
? Identify a molecule that belongs to the group.
By examining the effect of sequential application of the various symmetry operations in the group,
construct the group multiplication table.
Q7. a) How can group theory be used to determine whether an integral can be non-zero?
b) Use group theory to determine whether the following integrals are non-zero (use the tables of direct
products provided in the lecture handout).
i) the overlap integral between a p
x
orbital and a p
z
orbital in the point group C
2v
ii) the overlap integral between a p
x
orbital and a d
xz
orbital in the point group C
3v
iii) the overlap integral between a p
y
orbital and a d
z
2
orbital in the point group T
d
iv) the overlap integral between a p
z
orbital and a d
z
2
orbital in the point group D
2h
c) Which of the following electronic transitions are symmetry allowed?
i) a transition from a state ofA
1
symmetry to a state of E
1
symmetry excited by z-polarised light
in a molecule belonging to the point group C
5v
.
57
ii) a transition from a state of A
1g
symmetry to a state of A
2u
symmetry excited by z-polarised
light in a molecule belonging to the point group D
∞h
.
iii) a transition from a state of B
2
symmetry to a state of B
1
symmetry excited by y-polarised
light in a molecule belonging to the point group C
2v
.
Q8. Consider the hydronium ion H
3
O
+
. This ion has a pyramidal structure with one HOH bond angle smaller
than the other two, and belongs to the point group C
S
.
a) Using a basis set consisting of a 1s orbital on each H atom and 2s, 2p
x
, 2p
y
and 2p
z
orbitals on
the O atom (i.e. (s
O
,p
x
,p
y
,p
z
,s
1
,s
2
,s
3
)), construct a matrix representation.
b) What are the characters of each of the matrix representatives?
c) What are the irreps spanned by the basis?
d) Use the basis to construct a set of SALCs.
e) Write down the general form of the molecular orbitals of H
3
O
+
.
Q9. Consider the chlorobenzene molecule C
6
H
5
Cl.
a) What is the molecular point group?
b) Use a basis made up of a p orbital on each carbon atom (pointing perpendicular to the benzene
ring) to construct the molecular orbitals using the following steps:
i) determine the character of each symmetry operation
ii) determine the irreps spanned by the basis
iii) construct a set of SALCs and take linear combinations to form the molecular
orbitals of each symmetry species.
Q10. a) Use the 3N Cartesian basis and the character table for the C
3v
point group to determine the
symmetries of the vibrational modes of NH
3
.
b) Use a basis of internal coordinates to determine the symmetries of the stretching vibrations only.
Hence classify each of the vibrational modes found in a) as a bending or a stretching vibration.
c) Construct SALCs using the internal coordinate basis to determine the atomic displacements associated
with each stretching mode. Draw each mode, and label it as a symmetric or asymmetric stretching
vibration. It is quite complicated to use the 3N Cartesian basis to construct SALCs in this case
(though you are welcome to try). What do you think the A
1
bending vibration looks like? Identify the
A
1
and E bending vibrations as symmetric or antisymmetric.
d) Which vibrational modes could be excited by i) a one-photon process ii) a two-photon process? What
are the polarisations of the photons involved in each case?
