
Page 1
Introduction
This document describes the command-line tools included with SMRT Link
v8.0. These tools are for use by bioinformaticians working with secondary
analysis results.
• The command-line tools are located in the $SMRT_ROOT/smrtlink/
smrtcmds/bin
subdirectory.
Installation
The command-line tools are installed as an integral component of the SMRT
Link software. For installation details, see SMRT Link Software
Installation (v8.0).
• To install only the command-line tools, use the --smrttools-only
option with the installation command, whether for a new installation or
an upgrade. Examples:
smrtlink-*.run --rootdir smrtlink --smrttools-only
smrtlink-*.run --rootdir smrtlink --smrttools-only --upgrade
Pacific Biosciences Command-Line Tools
Following is information on the Pacific Biosciences-supplied command-line
tools included in the installation. Third-party tools installed are described at
the end of the document.
Tool Description
bam2fasta/
bam2fastq
Converts PacBio
®
BAM files into gzipped FASTA and FASTQ files.
See “bam2fasta/bam2fastq” on page 2.
bamsieve Generates a subset of a BAM or PacBio Data Set file based on either a
whitelist of hole numbers, or a percentage of reads to be randomly selected.
See “bamsieve” on page 3.
blasr Aligns long reads against a reference sequence. See “blasr” on page 5.
ccs Calculates consensus sequences from multiple “passes” around a circularized
single DNA molecule (SMRTbell
®
template). See “ccs” on page 10.
dataset Creates, opens, manipulates and writes Data Set XML files.
See “dataset” on page 15.
Demultiplex
Barcodes
Identifies barcode sequences in PacBio single-molecule sequencing data. See
“Demultiplex Barcodes” on page 21.
gcpp Variant-calling tool which provides several variant-calling algorithms for PacBio
sequencing data. See
“gcpp” on page 32.
ipdSummary Detects DNA base-modifications from kinetic signatures.
See “ipdSummary” on page 34.
SMRT
®
Tools Reference Guide

Page 2
bam2fasta/
bam2fastq
The bam2fastx tools convert PacBio BAM files into gzipped FASTA and
FASTQ files, including demultiplexing of barcoded data.
Usage
Both tools have an identical interface and take BAM and/or Data Set files
as input.
Examples
bam2fasta -o projectName m54008_160330_053509.subreads.bam
bam2fastq -o myEcoliRuns m54008_160330_053509.subreads.bam
m54008_160331_235636.subreads.bam
isoseq3 Characterizes full-length transcripts and generates full-length transcript
isoforms, eliminating the need for computational reconstruction.
See “isoseq3” on page 38.
juliet A general-purpose minor variant caller that identifies and phases minor single
nucleotide substitution variants in complex populations.
See “juliet” on page 41.
laa Finds phased consensus sequences from a pooled set of amplicons
sequenced with Pacific Biosciences’ SMRT technology. See
“laa” on page 49.
motifMaker Identifies motifs associated with DNA modifications in prokaryotic genomes.
See “motifMaker” on page 55.
pbalign Aligns PacBio reads to reference sequences; filters aligned reads according to
user-specified filtering criteria; and converts the output to PacBio BAM, SAM,
or PacBio DataSet format. See
“pbalign” on page 57.
pbcromwell Pacific Biosciences’ wrapper for the cromwell scientific workflow engine
used to power SMRT Link. For details on how to use pbcromwell to run
workflows, see
“pbcromwell” on page 60.
pbdagcon Implements DAGCon (Directed Acyclic Graph Consensus); a sequence
consensus algorithm based on using directed acyclic graphs to encode
multiple sequence alignments. See
“pbdagcon” on page 63.
pbindex Creates an index file that enables random access to PacBio-specific data in
BAM files. See
“pbindex” on page 64.
pbmm2 Aligns PacBio reads to reference sequences. A SMRT wrapper for minimap2,
and the successor to blasr and pbalign. See
“pbmm2” on page 65.
pbservice Performs a variety of useful tasks within SMRT Link.
See “pbservice” on page 71.
pbsv Structural variant caller for PacBio reads. See “pbsv” on page 75.
pbvalidate Validates that files produced by PacBio software are compliant with Pacific
Biosciences’ own internal specifications. See
“pbvalidate” on page 79.
sawriter Generates a suffix array file from an input FASTA file.
See “sawriter” on page 81.
summarizeModifica
tions
Generates a GFF summary file from the output of base modification analysis
combined with the coverage summary GFF generated by resequencing
pipelines. See
“summarize Modifications” on page 82.
Tool Description

Page 3
bam2fasta -o myHumanGenomem54012_160401_000001.subreadset.xml
Input Files
• One or more *.bam files
• *.subreadset.xml file (Data Set file)
Output Files
• *.fasta.gz
• *.fastq.gz
bamsieve
The bamsieve tool creates a subset of a BAM or PacBio Data Set file
based on either a whitelist of hole numbers, or a percentage of reads to be
randomly selected, while keeping all subreads within a read together.
Although
bamsieve is BAM-centric, it has some support for dataset XML
and will propagate metadata, as well as scraps BAM files in the special
case of SubreadSets.
bamsieve is useful for generating minimal test Data
Sets containing a handful of reads.
bamsieve operates in two modes: whitelist/blacklist mode where the
ZMWs to keep or discard are explicitly specified, or percentage/count
mode, where a fraction of the ZMWs is randomly selected.
ZMWs may be whitelisted or blacklisted in one of several ways:
• As a comma-separated list on the command line.
• As a flat text file, one ZMW per line.
• As another PacBio BAM or Data Set of any type.
Usage
bamsieve [-h] [--version] [--log-file LOG_FILE]
[--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL} | --debug | --quiet
| -v]
[--show-zmws] [--whitelist WHITELIST] [--blacklist BLACKLIST]
[--percentage PERCENTAGE] [-n COUNT] [-s SEED]
[--ignore-metadata][--barcodes]
input_bam [output_bam]
Required Description
input_bam The name of the input BAM file or Data Set from which reads will be read.
output_bam The name of the output BAM file or Data Set where filtered reads will be written to.
(Default = None)
Options Description
-h, --help Displays help information and exits.
--version Displays program version number and exits.
--log-file LOG_FILE Writes the log to file. (Default = None, writes to stdout.)

Page 4
Examples
Pulling out two ZMWs from a BAM file:
$ bamsieve --whitelist 111111,222222 full.subreads.bam sample.subreads.bam
Pulling out two ZMWs from a Data Set file:
$ bamsieve --whitelist 111111,222222 full.subreadset.xml sample.subreadset.xml
Using a text whitelist:
$ bamsieve --whitelist zmws.txt full.subreads.bam sample.subreads.bam
Using another BAM or Data Set as a whitelist:
$ bamsieve --whitelist mapped.alignmentset.xml full.subreads.bam mappable.subreads.bam
Generating a whitelist from a Data Set:
$ bamsieve --show-zmws mapped.alignmentset.xml > mapped_zmws.txt
Anonymizing a Data Set:
$ bamsieve --whitelist zmws.txt --ignore-metadata --anonymize full.subreadset.xml
anonymous_sample.subreadset.xml
Removing a read:
--log-level Specifies the log level; values are [DEBUG, INFO, WARNING, ERROR, CRITICAL].
(Default = WARNING)
--debug Alias for setting the log level to DEBUG. (Default = False)
--quiet Alias for setting the log level to CRITICAL to suppress output. (Default = False)
-v, --verbose Sets the verbosity level. (Default = NONE)
--show-zmws Prints a list of ZMWs and exits. (Default = False)
--whitelist WHITELIST Specifies the ZMWs to include in the output. This can be a comma-separated list
of ZMWs, or a file containing a list of ZMWs (one hole number per line), or a BAM/
Data Set file. (Default = NONE)
--blacklist BLACKLIST Specifies the ZMWs to exclude from the output. This can be a comma-separated
list of ZMWs, or a file containing a list of ZMWs (one hole number per line), or a
BAM/Data Set file that specifies ZMWs. (Default = NONE)
--percentage PERCENTAGE Specifies a percentage of a SMRT Cell to recover (Range = 1-100) rather than a
specific list of reads. (Default = NONE)
-n COUNT, --count COUNT Specifies a specific number of ZMWs picked at random to recover. (Default =
NONE)
-s SEED, --seed SEED Specifies a random seed for selecting a percentage of reads. (Default = NONE)
--ignore-metadata Discard the input Data Set metadata. (Default = False)
--barcodes Specifies that the whitelist or blacklist contains barcode indices instead of ZMW
numbers. (Default = False)
Options Description

Page 5
$ bamsieve --blacklist 111111 full.subreadset.xml filtered.subreadset.xml
Selecting 0.1% of reads:
$ bamsieve --percentage 0.1 full.subreads.bam random_sample.subreads.bam
Selecting a different 0.1% of reads:
$ bamsieve --percentage 0.1 --seed 98765 full.subreads.bam random_sample.subreads.bam
Selecting just two ZMWs/reads at random:
$ bamsieve --count 2 full.subreads.bam two_reads.subreads.bam
Selecting by barcode:
$ bamsieve --barcodes --whitelist 4,7 full.subreads.bam two_barcodes.subreads.bam
Generating a tiny BAM file that contains only mappable reads:
$ bamsieve --whitelist mapped.subreads.bam full.subreads.bam mappable.subreads.bam
$ bamsieve --count 4 mappable.subreads.bam tiny.subreads.bam
Splitting a Data Set into two halves:
$ bamsieve --percentage 50 full.subreadset.xml split.1of2.subreadset.xml
$ bamsieve --blacklist split.1of2.subreadset.xml full.subreadset.xml
split.2of2.subreadset.xml
Extracting Unmapped Reads:
$ bamsieve --blacklist mapped.alignmentset.xml movie.subreadset.xml
unmapped.subreadset.xml
blasr
The blasr tool aligns long reads against a reference sequence, possibly a
multi-contig reference.
blasr maps reads to genomes by finding the highest scoring local
alignment or set of local alignments between the read and the genome.
The initial set of candidate alignments is found by querying a rapidly-
searched precomputed index of the reference genome, and then refining
until only high-scoring alignments are kept. The base assignment in
alignments is optimized and scored using all available quality information,
such as insertion and deletion quality values.
Because alignment approximates an exhaustive search, alignment
significance is computed by comparing optimal alignment score to the
distribution of all other significant alignment scores.
Usage
blasr {subreads|ccs}.bam genome.fasta --bam --out aligned.bam [--options]
blasr {subreadset|consensusreadset}.xml genome.fasta --bam --out aligned.bam [--

Page 6
options]
blasr reads.fasta genome.fasta [--options]
Input Files
• {subreads|ccs}.bam is in PacBio BAM format, which is the native
Sequel
®
/Sequel II System output format of SMRT reads. PacBio BAM
files carry rich quality information (such as insertion, deletion, and
substitution quality values) needed for mapping, consensus calling and
variant detection. For the PacBio BAM format specifications, see
http://pacbiofileformats.readthedocs.io/en/5.1/BAM.html.
• {subreadset|consensusreadset}.xml is in PacBio Data Set format.
For the PacBio Data Set format specifications, see
http://pacbiofileformats.readthedocs.io/en/5.1/DataSet.html.
• reads.fasta: A multi-FASTA file of reads. While any FASTA file is
valid input,
bam or dataset files are preferable as they contain more
rich quality value information.
• genome.fasta: A FASTA file to which reads should map, usually
containing reference sequences.
Output Files
• aligned.bam: The pairwise alignments for each read, in PacBio BAM
format.
Input Options
Options for Aligning Output
Options Description
--sa suffixArrayFile Uses the suffix array sa for detecting matches between the reads and the reference.
(The suffix array is prepared by the sawriter program.)
--ctab tab Specifies a table of tuple counts used to estimate match significance, created by
printTupleCountTable. While it is quick to generate on the fly, if there are many
invocations of blasr, it is useful to precompute the ctab.
--regionTable table Specifies a read-region table in HDF format for masking portions of reads. This may
be a single table if there is just one input file, or a fofn (file-of-file names). When a
region table is specified, any region table inside the reads.plx.h5 or
reads.bax.h5 files is ignored. Note: This option works only with PacBio RS II
HDF5 files.
--noSplitSubreads Does not split subreads at adapters. This is typically only useful when the genome in
an unrolled version of a known template, and contains template-adapter-reverse-
template sequences. (Default = False)
Options Description
--bestn n Provides the top n alignments for the hit policy to select from. (Default = 10)
--sam Writes output in SAM format.
--bam Writes output in PacBio BAM format.
--clipping Uses no/hard/soft clipping for SAM output. (Default = none)
--out file Writes output to file. (Default = terminal)
--unaligned file Output reads that are not aligned to file.

Page 7
Options for Anchoring Alignment Regions
• These options will have the greatest effects on speed and sensitivity.
--m t If not printing SAM, modifies the output of the alignment.
•t=0: Print blast-like output with |'s connecting matched nucleotides.
• 1: Print only a summary: Score and position.
• 2: Print in Compare.xml format.
• 3: Print in vulgar format (Deprecated).
• 4: Print a longer tabular version of the alignment.
• 5: Print in a machine-parsable format that is read by
compareSequences.py.
--noSortRefinedAlignments Once candidate alignments are generated and scored via sparse dynamic
programming, they are rescored using local alignment that accounts for different
error profiles. Resorting based on the local alignment may change the order in
which the hits are returned. (Default = False)
--allowAdjacentIndels Allows adjacent insertion or deletions. Otherwise, adjacent insertion and
deletions are merged into one operation. Using quality values to guide pairwise
alignments may dictate that the higher probability alignment contains adjacent
insertions or deletions. Tools such as GATK do not permit this and so they are
not reported by default.
--header Prints a header as the first line of the output file describing the contents of each
column.
--titleTable tab Builds a table of reference sequence titles. The reference sequences are
enumerated by row, 0,1,... The reference index is printed in alignment results
rather than the full reference name. This makes output concise, particularly
when very verbose titles exist in reference names. (Default = NULL)
--minPctSimilarity p Reports alignments only if they are greater than p percent identity. (Default = 0)
--holeNumbers LIST Aligns reads whose ZMW hole numbers are in LIST only.
LIST is a comma-delimited string of ranges, such as 1,2,3,10-13. This
option only works when reads are in base or pulse h5 format.
--hitPolicy policy
Specifies how blasr treats multiple hits:
• all: Reports all alignments.
• allbest: Reports all equally top-scoring alignments.
• random: Reports a single random alignment.
• randombest: Reports a single random alignment from multiple equally top-
scoring alignments.
• leftmost: Reports an alignment which has the best alignment score and
has the smallest mapping coordinates in any reference.
Options Description
Options Description
--minMatch m Specifies the minimum seed length. A higher value will speed up alignment,
but decrease sensitivity. (Default = 12)
--maxMatch m
--maxLCPLength m
Stops mapping a read to the genome when the LCP length reaches m. This is
useful when the query is part of the reference, for example when constructing
pairwise alignments for de novo assembly. (Both options work the same.)
--maxAnchorsPerPosition m Do not add anchors from a position if it matches to more than m locations in
the target.
--advanceExactMatches E Speeds up alignments with match -E fewer anchors. Rather than finding
anchors between the read and the genome at every position in the read, when
an anchor is found at position i in a read of length L, the next position in a
read to find an anchor is at i+L-E. Use this when aligning already assembled
contigs. (Default = 0)

Page 8
Options for Refining Hits
Options for Overlap/Dynamic Programming Alignments and
Pairwise Overlap for de novo Assembly
--nCandidates n Keeps up to n candidates for the best alignment. A large value will slow
mapping as the slower dynamic programming steps are applied to more
clusters of anchors - this can be a rate-limiting step when reads are very long.
(Default = 10)
--concordant Maps all subreads of a ZMW (hole) to where the longest full pass subread of
the ZMW aligned to. This requires using the region table and hq regions. This
option only works when reads are in base or pulse h5 format.
(Default = False)
--placeGapConsistently Produces alignments with gaps placed consistently for better variant calling.
See “Gaps When Aligning” on page 10 for details.
Options Description
Options Description
--refineConcordantAlignments Refines concordant alignments. This slightly increases alignment accuracy
at the cost of time. This option is omitted if –-concordant is not set to
True. (Default = False)
--sdpTupleSize K Uses matches of length K to speed dynamic programming alignments. This
option controls accuracy of assigning gaps in pairwise alignments once a
mapping has been found, rather than mapping sensitivity itself.
(Default = 11)
--scoreMatrix "score matrix
string"
Specifies an alternative score matrix for scoring FASTA reads. The matrix is
in the format
ACGTN
A abcde
C fghij
G klmno
T pqrst
N uvwxy
The values a...y should be input as a quoted space separated string: "a
b c ... y". Lower scores are better, so matches should be less than
mismatches; such as a,g,m,s = -5 (match), mismatch = 6.
--affineOpen value Sets the penalty for opening an affine alignment. (Default = 10)
--affineExtend a Changes affine (extension) gap penalty. Lower value allows more gaps.
(Default = 0)
Options Description
--useQuality Uses substitution/insertion/deletion/merge quality values to score gap and mismatch
penalties in pairwise alignments. As the insertion and deletion rates are much higher
than substitution, this makes many alignments favor an insertion/deletion over a
substitution. Naive consensus-calling methods will then often miss substitution
polymorphisms. Use this option when calling consensus using the Quiver method.
Note: When not using quality values to score alignments, there will be a lower
consensus accuracy in homopolymer regions. (Default = False)
--affineAlign Refines alignment using affine guided align. (Default = False)

Page 9
Options for Filtering Reads
Options for Parallel Alignment
Options for Subsampling Reads
Examples
To align reads from reads.bam to the ecoli_K12 genome, and output in
PacBio BAM format:
blasr reads.bam ecoli_K12.fasta --bam --out ecoli_aligned.bam
To use multiple threads:
blasr reads.bam ecoli_K12.fasta --bam --out ecoli_aligned.bam --proc 16
To include a larger minimal match, for faster but less sensitive alignments:
blasr reads.bam ecoli_K12.fasta --bam --out ecoli_aligned.bam --proc 16 –-minMatch 15
To produce alignments in a pairwise human-readable format:
blasr reads.bam ecoli_K12.fasta -m 0
To use a precomputed suffix array for faster startup:
sawriter hg19.fasta.sa hg19.fasta #First precompute the suffix array
blasr reads.bam hg19.fasta --sa hg19.fasta.sa
Options Description
--minReadLength l Ignores reads that have a full length less than l. Subreads may be shorter.
(Default = 50)
--minSubreadLength l Does not align subreads of length less than l. (Default = 0)
--minAlnLength Reports alignments only if their lengths are greater than this value. (Default = 0)
Options Description
--nproc N Aligns using N processes. All large data structures such as the suffix array and tuple
count table are shared. (Default = 1)
--start S Index of the first read to begin aligning. This is useful when multiple instances are
running on the same data; for example when on a multi-rack cluster. (Default = 0)
--stride S Aligns one read every S reads. (Default = 1)
Options Description
--subsample p Proportion p of reads to randomly subsample and align; expressed as a decimal.
(Default = 0)
--help Displays help information and exits.
--version Displays version information using the format MajorVersion.Subversion.SHA1
(Example: 5.3.abcd123) and exits.

Page 10
Gaps When Aligning
By default, blasr places gap inconsistently when aligning a sequence
and its reverse-complement sequence. It is preferable to place gap
consistently to call a consensus sequence from multiple alignments or call
single nucleotide variants (SNPs), as the output alignments will make it
easier for variant callers to call variants.
Example:
REF : TTTTTTAAACCCC
READ1: TTTTTTACCCC
READ2: GGGGTAAAAAA
where READ1 and READ2 are reverse-complementary to each other.
In the following alignments, gaps are placed inconsistently:
REF : TTTTTTAAACCCC
READ1 : TTTTTTA--CCCC
RevComp(READ2): TTTTTT--ACCCC
In the following alignments, gaps are placed consistently, with
--placeGapsConsistently specified:
REF : TTTTTTAAACCCC
READ1 : TTTTTTA--CCCC
RevComp(READ2): TTTTTTA--CCCC
To produce alignments with gaps placed consistently for better variant
calling, use the
--placeGapConsistently option:
blasr query.bam target.fasta --out outfile.bam --bam -–placeGapConsistently
ccs
Circular Consensus Sequencing (CCS) calculates consensus sequences
from multiple “passes” around a circularized single DNA molecule
(SMRTbell
®
template). CCS uses the Arrow framework to achieve optimal
consensus results given the number of passes available.

Page 11
Input Files
• One .subreads.bam file containing the subreads for each SMRTbell
®
template sequenced.
Output Files
• A BAM file with one entry for each consensus sequence derived from a
ZMW. BAM is a general file format for storing sequence data, which is
described fully by the SAM/BAM working group. The CCS output
format is a version of this general format, where the consensus
sequence is represented by the "Query Sequence". Several tags were
added to provide additional meta information. An example BAM entry
for a consensus as seen by
samtools is shown below.
m141008_060349_42194_c100704972550000001823137703241586_s1_p0/63/ccs4*0255
**00CCCGGGGATCCTCTAGAATGC~~~~~~~~~~~~~~~~~~~~~RG:Z:83ba013f np:i:35 rq:f:0.999682
sn:B:f,11.3175,6.64119,11.6261,14.5199 zm:i:63
Following are some of the common fields contained in the output BAM file:
Field Description
Query Name Movie Name / ZMW # /ccs
FLAG Required by the format but meaningless in this context. Always set to 4 to indicate the
read is unmapped.
Reference Name Required by the format but meaningless in this context. Always set to *.
Mapping Start Required by the format but meaningless in this context. Always set to 0.
Mapping Quality Required by the format but meaningless in this context. Always set to 255.
CIGAR Required by the format but meaningless in this context. Always set to *.
RNEXT Required by the format but meaningless in this context. Always set to *.
PNEXT Required by the format but meaningless in this context. Always set to 0.
TLEN Required by the format but meaningless in this context. Always set to 0.

Page 12
Usage
ccs [OPTIONS] INPUT OUTPUT
Example
ccs --minlength 100 myData.subreads.bam myResult.bam
Consensus Sequence The consensus sequence generated.
Quality Values The per-base parametric quality metric. For details see “Interpreting QUAL Values” on
page 13.
RG Tag The read group identifier.
bc Tag A 2-entry array of upstream-provided barcode calls for this ZMW.
bq Tag The quality of the barcode call. (Optional: Depends on barcoded inputs.)
np Tag The number of full passes that went into the subread. (Optional: Depends on barcoded
inputs.)
rq Tag The predicted read quality.
t2 Tag The time (in seconds) spent aligning subreads to the draft consensus, prior to polishing.
t3 Tag The time (in seconds) spent polishing the draft consensus, not counting retries.
zm Tag The ZMW hole number.
Field Description
Required Description
Input File Name The name of a single subreads.bam or a subreadset.xml file to be processed.
(Example = myData.subreads.bam)
Output File Name The name of the output BAM file; comes after all other options listed. Valid output
files are the BAM and the Dataset .xml formats. (Example = myResult.bam)
Options Description
--version Prints the version number.
--report-file Contains a result tally of the outcomes for all ZMWs that were processed. If no file
name is given, the report is output to the file ccs_report.txt In addition to the
count of successfully-produced consensus sequences, this file lists how many
ZMWs failed various data quality filters (SNR too low, not enough full passes, and
so on) and is useful for diagnosing unexpected drops in yield.
--min-snr Removes data that is likely to contain deletions. SNR is a measure of the strength
of signal for all 4 channels (A, C, G, T) used to detect base pair incorporation. This
value sets the threshold for minimum required SNR for any of the four channels.
Data with SNR < 2.5 is typically considered lower quality. (Default = 2.5)
--min-length Specifies the minimum length requirement for the minimum length of the draft
consensus to be used for further polishing. If the targeted template is known to be a
particular size range, this can filter out alternative DNA templates. (Default = 10)
--max-length Specifies the maximum length requirement for the maximum length of the draft
consensus to be used for further polishing. For robust results while avoiding
unnecessary computation on unusual data, set to ~20% above the largest expected
insert size. (Default = 50000)

Page 13
Interpreting QUAL Values
The QUAL value of a read is a measure of the posterior likelihood of an
error at a particular position. Increasing QUAL values are associated with
a decreasing probability of error. For indels and homopolymers, there is
ambiguity as to which QUAL value is associated with the error probability.
Shown below are different types of alignment errors, with a
* indicating
which sequence BP should be associated with the alignment error.
Mismatch
*
ccs: ACGTATA
ref: ACATATA
Deletion
*
ccs: AC-TATA
ref: ACATATA
--min-passes Specifies the minimum number of passes for a ZMW to be emitted. This is the
number of full passes. Full passes must have an adapter hit before and after the
insert sequence and so do not include any partial passes at the start and end of the
sequencing reaction. It is computed as the number of passes mode across all
windows. (Default = 3)
--min-rq Specifies the minimum predicted accuracy of a read. ccs generates an accuracy
prediction for each read, defined as the expected percentage of matches in an
alignment of the consensus sequence to the true read. A value of 0.99 indicates
that only reads expected to be 99% accurate are emitted. (Default = 0.99)
--num-threads Specifies how many threads to use while processing. By default, ccs will use as
many threads as there are available cores to minimize processing time, but fewer
threads can be specified here.
--log-file The name of a log file to use. If none is given, the logging information is printed to
STDERR. (Example: mylog.txt)
--log-level Specifies verbosity of log data to produce. By setting --logLevel=DEBUG, you can
obtain detailed information on what ZMWs were dropped during processing, as well
as any errors which may have appeared. (Default = INFO)
--skip-polish After constructing the draft consensus, do not proceed with the polishing steps.
This is significantly faster, but generates less accurate data with no RQ or QUAL
values associated with each base.
--by-strand Separately generates a consensus sequence from the forward and reverse strands.
Useful for identifying heteroduplexes formed during sample preparation.
--chunk Operates on a single chunk. Format i/N, where i in [1,N]. Examples: 3/24 or 9/9.
--max-chunks Determines the maximum number of chunks, given an input file.
--modelPath Specifies the path to a model file or directory containing model files.
--modelSpec Specifies the name of the chemistry or model to use, overriding the default
selection.
Options Description

Page 14
Insertion
*
ccs: ACGTATA
ref: AC-TATA
Homopolymer Insertion or Deletion
Indels should always be left-aligned, and the error probability is only given
for the first base in a homopolymer.
* *
ccs: ACGGGGTATA ccs: AC-GGGTATA
ref: AC-GGGTATA ref: ACGGGGTATA
CCS Yield Report
The CCS Report specifies the number of ZMWs that successfully
produced consensus sequences, as well as a count of how many ZMWs
did not produce a consensus sequence for various reasons. The entries in
this report, as well as parameters used to increase or decrease the
number of ZMWs that pass various filters, are shown in the table below.
The first part is a summary of inputs and outputs:
The second part explains in details the exclusive ZMW count for (C),
those ZMWs that were filtered:
ZMW Results
Parameters Affecting
Results
Description
ZMWs input (A) None The number of input ZMWs.
ZMWs generating CCS (B) All custom processing settings The number of CCS reads successfully produced on
the first attempt, using the fast windowed approach.
ZMWs filtered (C) All custom processing settings The number of ZMWs reads that failed producing a
CCS read.
ZMW Results
Parameters Affecting
Results
Description
No usable subreads --minReadScore,
--minLength,
--maxLength
The ZMW had no usable subreads. Either there were no
subreads, or all subreads had lengths outside the range
<50% or >200% of the median subread length.
Below SNR threshold --min-snr The ZMW had at least one channel's SNR below the
minimum threshold.
Lacking full passes --min-passes There were not enough subreads that had an adapter at
the start and end of the subread (a "full pass").
Heteroduplexes None The SMRTbell contains a heteroduplex. In this case, it is
not clear what the consensus should be and so the ZMW is
dropped.
Min coverage violation None The ZMW is damaged on one strand and cannot be
polished reliably.

Page 15
dataset
The dataset tool creates, opens, manipulates and writes Data Set XML
files. The commands allow you to perform operations on the various types
of data held by a Data Set XML: Merge, split, write, and so on.
Usage
dataset [-h] [--version] [--log-file LOG_FILE]
[--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL} | --debug | --quiet | -v]
[--strict] [--skipCounts]
{create,filter,merge,split,validate,summarize,consolidate,loadstats,newuuid,loadmetada
ta,copyto,absolutize,relativize}
create Command: Create an XML file from a fofn (file-of-file names) or
BAM file. Possible types:
SubreadSet, AlignmentSet, ReferenceSet,
Draft generation error None Subreads do not match the generated draft sequence,
even after multiple tries.
Draft above --max-
length
--max-length The draft sequence was above the maximum length
threshold.
Draft below --min-
length
--min-length The draft sequence was below the minimum length
threshold.
Lacking usable
subreads
None Too many subreads were dropped while polishing
CCS did not converge None The consensus sequence did not converge after the
maximum number of allowed rounds of polishing.
CCS below minimum
predicted accuracy
--min-rq Each CCS read has a predicted level of accuracy
associated with it. Reads that are below the minimum
specified threshold are removed.
Unknown error during
processing
None These should not occur.
ZMW Results
Parameters Affecting
Results
Description
Options Description
-h, --help Displays help information and exits.
<Command> -h Displays help for a specific command.
-v, --version Displays program version number and exits.
--log-file LOG_FILE Writes the log to file. (Default = None, writes to stdout.)
--log-level Specifies the log level; values are [DEBUG, INFO, WARNING, ERROR,
CRITICAL]. (Default = INFO)
--debug Alias for setting the log level to DEBUG. (Default = False)
--quiet Alias for setting the log level to CRITICAL to suppress output.
(Default = False)
-v Sets the verbosity level. (Default = NONE)
--strict Turns on strict tests and display all errors. (Default = False)
--skipCounts Skips updating NumRecords and TotalLength counts.
(Default = False)

Page 16
HdfSubreadSet, BarcodeSet, ConsensusAlignmentSet,
ConsensusReadSet, ContigSet
.
dataset create [-h] [--type DSTYPE] [--name DSNAME] [--generateIndices]
[--metadata METADATA] [--novalidate] [--relative]
outfile infile [infile ...]
Example
The following example shows how to use the dataset create command
to create a barcode file:
$ dataset create --generateIndices --name my_barcodes --type BarcodeSet
my_barcodes.barcodeset.xml my_barcodes.fasta
filter Command: Filter an XML file using filters and threshold values.
• Suggested filters: accuracy, bc, bcf, bcq, bcr, bq, cx, length, movie,
n_subreads, pos, qend, qname, qstart, readstart, rname, rq, tend,
tstart, zm.
• More resource-intensive filter: [qs]
Note: Multiple filters with different names are ANDed together. Multiple
filters with the same name are ORed together, duplicating existing
requirements.
dataset filter [-h] infile outfile filters [filters ...]
Required Description
outfile The name of the XML file to create.
infile The fofn (file-of-file-names) or BAM file(s) to convert into an XML file.
Options Description
--type DSTYPE Specifies the type of XML file to create. (Default = NONE)
--name DSNAME The name of the new Data Set XML file.
--generateIndices Generates index files (.pbi and .bai for BAM, .fai for FASTA). Requires
samtools/pysam and pbindex. (Default = FALSE)
--metadata METADATA A metadata.xml file (or Data Set XML) to supply metadata.
(Default = NONE)
--novalidate Specifies not to validate the resulting XML. Leaves the paths as they are.
--relative Makes the included paths relative instead of absolute. This is not
compatible with --novalidate.
Required Description
infile The name of the XML file to filter.
outfile The name of the output filtered XML file.

Page 17
merge Command: Combine XML files.
dataset merge [-h] outfile infiles [infiles ...]
split Command: Split a Data Set XML file.
dataset split [-h] [--contigs] [--barcodes] [--zmws] [--byRefLength]
[--noCounts] [--chunks CHUNKS] [--maxChunks MAXCHUNKS]
[--targetSize TARGETSIZE] [--breakContigs]
[--subdatasets] [--outdir
infile [outfiles...]
validate Command: Validate XML and ResourceId files. (This is an
internal testing functionality that may be useful.)
Note: This command requires that pyxb (not distributed with SMRT Link)
be installed. If not installed,
validate simply checks that the files pointed
to in
ResourceIds exist.
filters The values to filter on. (Example: rq>0.85)
Required Description
Required Description
infiles The names of the XML files to merge.
outfile The name of the output XML file.
Required Description
infile The name of the XML file to split.
Options Description
outfiles The names of the resulting XML files.
--contigs Splits the XML file based on contigs. (Default = FALSE)
--barcodes Splits the XML file based on barcodes. (Default = FALSE)
--zmws Splits the XML file based on ZMWs. (Default = FALSE)
--byRefLength Splits contigs by contig length. (Default = TRUE)
--noCounts Updates the Data Set counts after the split. (Default = FALSE)
--chunks x Splits contigs into x total windows. (Default = 0)
--maxChunks x Splits the contig list into at most x groups. (Default = 0)
--targetSize x Specifies the minimum number of records per chunk. (Default = 5000)
--breakContigs Breaks contigs to get closer to maxCounts. (Default = False)
--subdatasets Splits the XML file based on subdatasets. (Default = False)
--outdir OUTDIR Specifies an output directory for the resulting XML files.
(Default = <in-place>, not the current working directory.)

Page 18
dataset validate [-h] [--skipFiles] infile
summarize Command: Summarize a Data Set XML file.
dataset summarize [-h] infile
consolidate Command: Consolidate XML files.
dataset consolidate [-h] [--numFiles NUMFILES] [--noTmp]
infile datafile xmlfile
loadstats Command: Load an sts.xml file containing pipeline statistics
into a Data Set XML file.
dataset loadstats [-h] [--outfile OUTFILE] infile statsfile
Required Description
infile The name of the XML file to validate.
Options Description
--skipFiles Skips validating external resources. (Default = False)
Required Description
infile The name of the XML file to summarize.
Required Description
infile The name of the XML file to consolidate.
datafile The name of the resulting data file.
xmlfile The name of the resulting XML file.
Options Description
--numFiles x Specifies the number of data files to produce. (Default = 1)
--noTmp Do not copy to a temporary location to ensure local disk use.
(Default = False)
Required Description
infile The name of the Data Set XML file to modify.
statsfile The name of the .sts.xml file to load.
Options Description
--outfile OUTFILE The name of the XML file to output. (Default = None)

Page 19
newuuid Command: Refresh a Data Set's Unique ID.
dataset newuuid [-h] [--random] infile
loadmetadata Command: Load a .metadata.xml file into a Data Set
XML file.
dataset loadmetadata [-h] [--outfile OUTFILE] infile metadata
copyto Command: Copy a Data Set and resources to a new location.
dataset copyto [-h] [--relative] infile outdir
absolutize Command: Make the paths in an XML file absolute.
dataset absolutize [-h] [--outdir OUTDIR] infile
Required Description
infile The name of the XML file to refresh.
Options Description
--random Generates a random UUID, instead of a hash. (Default = False)
Required Description
infile The name of the Data Set XML file to modify.
metadata The .metadata.xml file to load, or Data Set to borrow from.
Options Description
--outfile OUTFILE Specifies the XML file to output. (Default = None)
Required Description
infile The name of the XML file to copy.
outdir The directory to copy to.
Options Description
--relative Makes the included paths relative instead of absolute. (Default = False)
Required Description
infile The name of the XML file whose paths should be absolute.
Options Description
--outdir OUTDIR Specifies an optional output directory. (Default = None)

Page 20
relativize Command: Make the paths in an XML file relative.
dataset relativize [-h] infile
Example - Filter Reads
To filter one or more BAM file’s worth of subreads, aligned or otherwise,
and then place them into a single BAM file:
# usage: dataset filter <in_fn.xml> <out_fn.xml> <filters>
dataset filter in_fn.subreadset.xml filtered_fn.subreadset.xml 'rq>0.85'
# usage: dataset consolidate <in_fn.xml> <out_data_fn.bam> <out_fn.xml>
dataset consolidate filtered_fn.subreadset.xml consolidate.subreads.bam
out_fn.subreadset.xml
The filtered Data Set and the consolidated Data Set should be read-for-
read equivalent when used with SMRT
®
Analysis software.
Example - Resequencing Pipeline
• Align two movie’s worth of subreads in two SubreadSets to a
reference.
• Merge the subreads together.
• Split the subreads into Data Set chunks by contig.
• Process using gcpp on a chunkwise basis (in parallel).
1. Align each movie to the reference, producing a Data Set with one BAM
file for each execution:
pbalign movie1.subreadset.xml referenceset.xml movie1.alignmentset.xml
pbalign movie2.subreadset.xml referenceset.xml movie2.alignmentset.xml
2. Merge the files into a FOFN-like Data Set; BAMs are not touched:
# dataset merge <out_fn> <in_fn> [<in_fn> <in_fn> ...]
dataset merge merged.alignmentset.xml movie1.alignmentset.xml movie2.alignmentset.xml
3. Split the Data Set into chunks by contig name; BAMs are not touched:
– Note that supplying output files splits the Data Set into that many
output files (up to the number of contigs), with multiple contigs per
file.
– Not supplying output files splits the Data Set into one output file per
contig, named automatically.
– Specifying a number of chunks instead will produce that many files,
with contig or even subcontig (reference window) splitting.
dataset split --contigs --chunks 8 merged.alignmentset.xml
Required Description
infile The name of the XML file whose paths should be relative.

Page 21
4. Process the chunks:
gcpp --reference referenceset.xml --output
chunk1consensus.fasta,chunk1consensus.fastq,chunk1consensus.vcf,chunk1consensus.gff
chunk1contigs.alignmentset.xml
The chunking works by duplicating the original merged Data Set (no BAM
duplication) and adding filters to each duplicate such that only reads
belonging to the appropriate contigs are emitted. The contigs are
distributed among the output files in such a way that the total number of
records per chunk is about even.
Demultiplex
Barcodes
The Demultiplex Barcodes application identifies barcode sequences in
PacBio single-molecule sequencing data. It replaced
pbbarcode and
bam2bam for demultiplexing, starting with SMRT
®
Analysis v5.1.0.
Demultiplex Barcodes can demultiplex samples that have a unique per-
sample barcode pair and were pooled and sequenced on the same SMRT
Cell. There are four different methods for barcoding samples with PacBio
technology:
1. Sequence-specific primers
2. Barcoded universal primers
3. Barcoded adapters
4. Linear Barcoded Adapters for Probe-based Captures

Page 22
In addition, there are three different barcode library designs. As
Demultiplex Barcodes supports raw subread and CCS read
demultiplexing, the following terminology is based on the per (sub-) read
view.

Page 23
In the overview above, the input sequence is flanked by adapters on both
sides. The bases adjacent to an adapter are barcode regions. A read can
have up to two barcode regions, leading and trailing. Either or both adapt
-
ers can be missing and consequently the leading and/or trailing region is
not being identified.
For symmetric and tailed library designs, the same barcode is attached
to both sides of the insert sequence of interest. The only difference is the
orientation of the trailing barcode. For barcode identification, one read with
a single barcode region is sufficient.
For the asymmetric design, different barcodes are attached to the sides
of the insert sequence of interest. To identify the different barcodes, a read
with leading and trailing barcode regions is required.
Output barcode pairs are generated from the identified barcodes. The bar-
code names are combined using “--“, for example bc1002--bc1054. The
sort order is defined by the barcode indices, starting with the lowest.
Workflow
By default, Demultiplex Barcodes processes input reads grouped by
ZMW, except if the
--per-read option is used. All barcode regions along
the read are processed individually. The final per-ZMW result is a
summary over all barcode regions. Each ZMW is assigned to a pair of
selected barcodes from the provided set of candidate barcodes. Subreads
from the same ZMW will have the same barcode and barcode quality. For
a particular target barcode region, every barcode sequence gets aligned
as given and as reverse-complement, and higher scoring orientation is
chosen. This results in a list of scores over all candidate barcodes.
• If only same barcode pairs are of interest (symmetric/tailed), use the
--same option to filter out different barcode pairs.
• If only different barcode pairs are of interest (asymmetric), use the
--different option to require at least two barcodes to be read, and
remove pairs with the same barcode.

Page 24
Half Adapters
For an adapter call with only one barcode region, the high-quality region
finder cuts right through the adapter. The preceding or succeeding
subread was too short and was removed, or the sequencing reaction
started/stopped there. This is called a half adapter. Thus, there are also
1.5, 2.5, N+0.5 adapter calls.
ZMWs with half or only one adapter can be used to identify the same
barcode pairs; positive-predictive value might be reduced compared to
high adapter calls. For asymmetric designs with different barcodes in a
pair, at least a single full-pass read is required; this can be two adapters,
two half adapters, or a combination.
Usage:
• Any existing output files are overwritten after execution.
• Always use --peek-guess to remove spurious barcode hits.
Analysis of subread data:
lima movie.subreads.bam barcodes.fasta prefix.bam
lima movie.subreadset.xml barcodes.barcodeset.xml prefix.subreadset.xml
Analysis of CCS data:
lima --css movie.ccs.bam barcodes.fasta prefix.bam
lima --ccs movie.consensusreadset.xml barcodes.barcodeset.xml
prefix.consensusreadset.xml
If you do not need to import the demultiplexed data into SMRT Link, use
the
--no-pbi option to minimize memory consumption and run time.
Symmetric or Tailed options:
Raw: --same
CCS: --same --ccs
Asymmetric options:
Raw: --different
CCS: --different --ccs
Example Execution:
lima m54317_180718_075644.subreadset.xml \
Sequel_RSII_384_barcodes_v1.barcodeset.xml \
m54317_180718_075644.demux.subreadset.xml \
--different --peek-guess
Options Description
--same Retains only reads with the same barcodes on both ends of the insert
sequence, such as symmetric and tailed designs.
--different Retains only reads with different barcodes on both ends of the insert
sequence, asymmetric designs. Enforces --min-passes ≥ 1.

Page 25
--min-length n Omits reads with lengths below n base pairs after demultiplexing. ZMWs
with no reads passing are omitted. (Default = 50)
--max-input-length n Omits reads with lengths above n base pairs for scoring in the
demultiplexing step. (Default = 0, deactivated)
--min-score n Omits ZMWs with average barcode scores below n. A barcode score
measures the alignment between a barcode attached to a read and an
ideal barcode sequence, and is an indicator how well the chosen barcode
pair matches. It is normalized to a range between 0 (no hit) and 100 (a
perfect match).
(Default = 0, Pacific Biosciences recommends setting it to 26.)
--min-end-score n Specifies the minimum end barcode score threshold applied to the
individual leading and trailing ends. (Default = 0)
--min-passes n Omits ZMWs with less than n full passes, a read with a leading and trailing
adapter. (Default = 0, no full-pass needed) Example:
0 pass : insert - adapter - insert
1 pass : insert - adapter - INSERT - adapter - insert
2 passes: insert - adapter - INSERT - adapter - INSERT -
adapter - insert
--score-full-pass Uses only reads flanked by adapters on both sides (full-pass reads) for
barcode identification.
--min-ref-span Specifies the minimum reference span relative to the barcode length.
(Default = 0.5)
--per-read Scores and tags per subread, instead of per ZMW.
--ccs Sets defaults to -A 1 -B 4 -D 3 -I 3 -X 1.
--peek n Looks at the first n ZMWs of the input and return the mean. This lets you
test multiple test barcode.fasta files and see which set of barcodes
was used.
--guess n This performs demultiplexing twice. In the first iteration, all barcodes are
tested per ZMW. Afterwards, the barcode occurrences are counted and
their mean is tested against the threshold n; only those barcode pairs that
pass this threshold are used in the second iteration to produce the final
demultiplexed output. A prefix.lima.guess file shows the decision
process; --same is being respected.
--guess-min-count Specifies the minimum ZMW count to whitelist a barcode. This filter is
ANDed with the minimum barcode score specified by --guess.
(Default = 0)
--peek-guess Equivalent to the Infer Barcodes Used parameter option in SMRT Link.
Sets the following options:
--peek 50000 --guess 45 --guess-min-count 10.
Demultiplex Barcodes will run twice on the input data. For the first 50,000
ZMWs, it will guess the barcodes and store the mask of identified
barcodes. In the second run, the barcode mask is used to demultiplex all
ZMWs.
If combined with --ccs then the barcode score threshold is increased by
--guess 75.
--single-side Identifies barcodes in molecules that only have barcodes adjacent to one
adapter.
--window-size-mult
--window-size-bp
The candidate region size multiplier: barcode_length *
multiplier. (Default = 3)
Optionally, you can specify the region size in base pairs using
--window-size-bp. If set, --window-size-mult is ignored.
Options Description

Page 26
Input Files:
Input data in PacBio-enhanced BAM format is either:
• Sequence data - Unaligned subreads, directly from a Sequel/Sequel II
System, or
• Unaligned CCS reads, generated by CCS 2.
Note: To demultiplex PacBio RS II data, use SMRT Link or bax2bam to
convert
.h5 files to BAM format.
Barcodes are provided as a FASTA file or BarcodeSet file:
• One entry per barcode sequence.
• No duplicate sequences.
• All bases must be in upper-case.
• Orientation-agnostic (forward or reverse-complement, but not
reversed.)
Example:
>bc1000
CTCTACTTACTTACTG
--num-threads n Spawns n threads; 0 means use all available cores. This option also
controls the number of threads used for BAM and PBI compression.
(Default = 0)
--chunk-size n Specifies that each thread consumes n ZMWs per chunk for processing.
(Default = 10).
--no-bam Does not produce BAM output. Useful if only reports are of interest, as run
time is shorter.
--no-pbi Does not produce a .bam.pbi index file. The on-the-fly .bam.pbi file
generation buffers the output data. If you do not need a .bam.pbi index
file for SMRT Link import, use this option to decrease memory usage to a
minimum and shorten the run time.
--no-reports Does not produce any reports. Useful if only demultiplexed BAM files are
needed.
--dump-clips Outputs all clipped barcode regions generated to the
<prefix>.lima.clips file.
--dump-removed Outputs all records that did not pass the specified thresholds, or are
without barcodes, to the <prefix>.lima.removed.bam file.
--split-bam
--split-bam-named
Specifies that each barcode has its own BAM file called
prefix.idxBest-idxCombined.bam, such as prefix.0-0.bam.
Optionally ,--split-bam-named names the files by their barcode
names instead of their barcode indices.
--isoseq Removes primers as part of the Iso-Seq pipeline.
See “Demultiplexing Iso-Seq Data” on page 31 for details.
--bad-adapter-ratio n Specifies the maximum ratio of bad adapters. (Default = 0).
Options Description

Page 27
>bc1001
GTCGTATCATCATGTA
>bc1002
AATATACCTATCATTA
Note: Name barcodes using an alphabetic character prefix to avoid later
barcode name/index confusion.
Output Files:
Demultiplex Barcodes generates multiple output files by default, all
starting with the same prefix as the output file, using the suffixes
.bam,
.subreadset.xml, and .consensusreadset.xml. The report prefix is
lima. Example:
lima m54007_170702_064558.subreads.bam barcode.fasta /my/path/
m54007_170702_064558_demux.subreadset.xml
For all output files, the prefix is
/my/path/m54007_170702_064558_demux.
• <prefix>.bam: Contains clipped records, annotated with barcode
tags, that passed filters and respect the
--same option.
• <prefix>.lima.report: A tab-separated file describing each ZMW,
unfiltered. This is useful information for investigating the demultiplexing
process and the underlying data. A single row contains all reads from
a single ZMW. For
--per-read, each row contains one subread, and
ZMWs might span multiple rows.
• <prefix>.lima.summary: Lists how many ZMWs were filtered, how
many ZMWs are the same or different, and how many reads were
filtered.
(1)
ZMWs input (A) : 213120
ZMWs above all thresholds (B) : 176356 (83%)
ZMWs below any threshold (C) : 36764 (17%)
(2)
ZMW Marginals for (C) :
Below min length : 26 (0%)
Below min score : 0 (0%)
Below min end score : 5138 (13%)
Below min passes : 0 (0%)
Below min score lead : 11656 (32%)
Below min ref span : 3124 (8%)
Without adapter : 25094 (68%)
With bad adapter : 10349 (28%) <- Only with --bad-adapter-ratio
Undesired hybrids : xxx (xx%) <- Only with --peek-guess
Undesired same barcode pairs : xxx (xx%) <- Only with --different
Undesired diff barcode pairs : xxx (xx%) <- Only with --same
Undesired 5p--5p pairs : xxx (xx%) <- Only with --isoseq
Undesired 3p--3p pairs : xxx (xx%) <- Only with --isoseq
Undesired single side : xxx (xx%) <- Only with --isoseq
Undesired no hit : xxx (xx%) <- Only with --isoseq

Page 28
(3)
ZMWs for (B):
With same barcode : 162244 (92%)
With different barcodes : 14112 (8%)
Coefficient of correlation : 32.79%
(4)
ZMWs for (A):
Allow diff barcode pair : 157264 (74%)
Allow same barcode pair : 188026 (88%)
Bad adapter yield loss : 10112 (5%) <- Only with --bad-adapter-ratio
Bad adapter impurity : 10348 (5%) <- Only without --bad-adapter-ratio
(5)
Reads for (B):
Above length : 1278461 (100%)
Below length : 2787 (0%)
Explanation of each block:
1. Number of ZMWs that went into lima, how many ZMWs were passed
to the output file, and how many did not qualify.
2. For those ZMWs that did not qualify: The marginal counts of each filter.
(Filter are described in the Options table.)
When running with --peek-guess or similar manual option combina-
tion and different barcode pairs are found during peek, the full SMRT
Cell may contain low-abundant different barcode pairs that were identi
-
fied during peek individually, but not as a pair. Those unwanted bar-
code pairs are called hybrids.
3. For those ZMWs that passed: How many were flagged as having the
same or different barcode pair, as well as the coefficient of variation for
the barcode ZMW yield distribution in percent.
4. For all input ZMWs: How many allow calling the same or different bar-
code pair. This is a simplified version of how many ZMW have at least
one full pass to allow a different barcode pair call and how many
ZMWs have at least half an adapter, allowing the same barcode pair
call.
5. For those ZMWs that qualified: The number of reads that are above
and below the specified
--min-length threshold.
• <prefix>.lima.counts: A .tsv file listing the counts of each
observed barcode pair. Only passing ZMWs are counted. Example:
$ column -t prefix.lima.counts
IdxFirst IdxCombined IdxFirstNamed IdxCombinedNamed Counts MeanScore
0 0 bc1001 bc1001 1145 68
1 1 bc1002 bc1002 974 69
2 2 bc1003 bc1003 1087 68

Page 29
• <prefix>.lima.clips: Contains clipped barcode regions generated
using the
--dump-clips option. Example:
$ head -n 6 prefix.lima.clips
>m54007_170702_064558/4850602/6488_6512 bq:34 bc:11
CATGTCCCCTCAGTTAAGTTACAA
>m54007_170702_064558/4850602/6582_6605 bq:37 bc:11
TTTTGACTAACTGATACCAATAG
>m54007_170702_064558/4916040/4801_4816 bq:93 bc:10
• <prefix>.lima.removed.bam: Contains records that did not pass the
specified thresholds, or are without barcodes, using the option
--dump-removed.
lima does not generate a .pbi, nor Data Set for this file. This option
cannot be used with any splitting option.
• <prefix>.lima.guess: A .tsv file that describes the barcode
subsetting process activated using the
--peek and --guess options.
• One DataSet,.subreadset.xml, or .consensusreadset.xml file is
generated per output BAM file.
• .pbi: One PBI file is generated per output BAM file.
What is a universal spacer sequence and how does it affect
demultiplexing?
For library designs that include an identical sequence between adapter
and barcode, such as probe-based linear barcoded adapters samples,
Demultiplex Barcodes offers a special mode that is activated if it finds a
shared prefix sequence among all provided barcode sequences.
Example:
>custombc1
ACATGACTGTGACTATCTCACACATATCAGAGTGCG
>custombc2
ACATGACTGTGACTATCTCAACACACAGACTGTGAG
In this case, Demultiplex Barcodes detects the shared prefix
ACATGACTGTGACTATCTCA and removes it internally from all barcodes.
Subsequently, it increases the window size by the length
L of the prefix
sequence.
• If --window-size-bp N is used, the actual window size is L + N.
IdxFirst IdxCombined IdxFirstNamed IdxCombinedNamed NumZMWs MeanScore Picked
0 0 bc1001t bc1001t 1008 50 1
1 1 bc1002t bc1002t 1005 60 1
2 2 bc1003t bc1003t 5 24 0
3 3 bc1004t bc1004t 555 61 1

Page 30
• If --window-size-mult M is used, the actual window size is
(L + |bc|) * M.
Because the alignment is semi-global, a leading reference gap can be
added without any penalty to the barcode score.
What are bad adapters?
In the subreads.bam file, each subread has a context flag cx. The flag
specifies, among other things, whether a subread has flanking adapters,
before and/or after. Adapter-finding was improved and can also find
molecularly-missing adapters, or those obscured by a local decrease in
accuracy. This may lead to missing or obscured bases in the flanking
barcode. Such adapters are labelled "bad", as they don't align with the
adapter reference sequence(s). Regions flanking those bad adapters are
problematic, because they can fully or partially miss the barcode bases,
leading to wrong classification of the molecule.
lima can handle those
adapters by ignoring regions flanking bad adapters. For this,
lima
computes the ratio of number of bad adapters divided by number of all
adapters.
By default, --bad-adapter-ratio is set to 0 and does not perform any
filtering. In this mode, bad adapters are handled just like good adapters.
But the *.lima.summary file contains one row with the number of ZMWs
that have at least 25% bad adapters, but otherwise pass all other filters.
This metric can be used as a diagnostic to assess library preparation.
If --bad-adapter-ratio is set to non-zero positive (0,1), bad adapter
flanking barcode regions are treated as missing. If a ZMW has a higher
ratio of bad adapters than provided, the ZMW is filtered and consequently
removed from the output. The
*.lima.summary file contains two
additional rows.
With bad adapter : 10349 (28%)
Bad adapter yield loss : 10112 (5%)
The first row counts the number of ZMWs that have bad adapter ratios that
are too high; the percentage is with respect to the number of all ZMW not
passing. The second row counts the number of ZMWs that are removed
solely due to bad adapter ratios that are too high; the percentage is with
respect the number of all input ZMWs and consequently is the effective
yield loss caused by bad adapters.
If a ZMW has ~50% bad adapters, one side of the molecule is molecularly-
missing an adapter. For 100% bad adapter, both sides are missing
adapters. A lower than ~40% percentage indicates decreased local
accuracy during sequencing leading to adapter sequences not being
found. If a high percentage of ZMWs is molecularly-missing adapters, you
should improve library preparation.

Page 31
Demultiplexing Iso-Seq Data
Demultiplex Barcodes is used to identify and remove Iso-Seq cDNA
primers. If the Iso-Seq sample is barcoded, the barcodes should be
included as part of the primer. Note: To demultiplex Iso-Seq samples in the
SMRT Link (GUI), always choose the Iso-Seq Analysis or Iso-Seq
Analysis with Mapping applications, not the Demultiplex Barcodes
application. Only by using the command line can users use
lima with the
--isoseq option for demultiplexing Iso-Seq data.
The input Iso-Seq data format for demultiplexing is .ccs.bam. Users must
first generate a CCS BAM file for an Iso-Seq Data Set before running
lima. The recommended parameters for running CCS for Iso-Seq are
min-pass=1, min accuracy=0.8, and turning Polish to OFF.
1. Primer IDs must be specified using the suffix _5p to indicate 5’ cDNA
primers and the suffix
_3p to indicate 3’ cDNA primers. The 3’ cDNA
primer should not include the Ts and is written in reverse complement.
2. Below are two example primer sets. The first is unbarcoded, the sec-
ond has barcodes (shown in lower case) adjacent to the 3’ primer.
Example 1: The IsoSeq v2 primer set.
>NEB_5p
GCAATGAAGTCGCAGGGTTGGG
>Clontech_5p
AAGCAGTGGTATCAACGCAGAGTACATGGGG
>NEB_Clontech_3p
GTACTCTGCGTTGATACCACTGCTT
Example 2: 4 tissues were multiplexed using barcodes on the 3’ end
only.
>5p
AAGCAGTGGTATCAACGCAGAGTACATGGGG
>tissue1_3p
atgacgcatcgtctgaGTACTCTGCGTTGATACCACTGCTT
>tissue2_3p
gcagagtcatgtatagGTACTCTGCGTTGATACCACTGCTT
>tissue3_3p
gagtgctactctagtaGTACTCTGCGTTGATACCACTGCTT
>tissue4_3p
catgtactgatacacaGTACTCTGCGTTGATACCACTGCTT
3. Use the --isoseq mode. Note that this cannot be combined with the
--guess option.
4. The output will be only different pairs with a 5p and 3p combination:
demux.5p--tissue1_3p.bam
demux.5p--tissue2_3p.bam
The --isoseq parameter set is very conservative for removing any
spurious and ambiguous calls, and guarantees that only proper
asymmetric (barcoded) primer are used in downstream analyses. Good
libraries reach >75% CCS reads passing the Demultiplex Barcodes filters.

Page 32
gcpp
gcpp is a variant-calling tool provided by the GCpp package which provides
several variant-calling algorithms for PacBio sequencing data.
Usage
gcpp -j8 --algorithm=arrow \
-r lambdaNEB.fa \
-o variants.gff \
aligned_subreads.bam
This example requests variant-calling, using 8 worker processes and the
Arrow algorithm, taking input from the file
aligned_subreads.bam, using
the FASTA file
lambdaNEB.fa as the reference, and writing output to
variants.gff.
A particularly useful option is --referenceWindow/-w; which allows the
variant-calling to be performed exclusively on a window of the reference
genome.
Input Files
• A sorted file of reference-aligned reads in Pacific Biosciences’
standard BAM format.
• A FASTA file that follows the Pacific Biosciences FASTA file
convention.
Note: The --algorithm=arrow option requires that certain metrics be in
place in the input BAM file. It requires per-read SNR metrics, and the per-
base
PulseWidth metric for Sequel data.
The selected algorithm will stop with an error message if any features that
it requires are unavailable.
Output Files
Output files are specified as comma-separated arguments to the -o flag.
The file name extension provided to the
-o flag is meaningful, as it
determines the output file format. For example:
gcpp aligned_subreads.bam -r lambda.fa -o myVariants.gff,myConsensus.fasta
will read input from aligned_subreads.bam, using the reference
lambda.fa, and send variant call output to the file myVariants.gff, and
consensus output to
myConsensus.fasta.
The file formats currently supported (using extensions) are:
• .gff: PacBio GFFv3 variants format; convertible to BED.
• .vcf: VCF 4.2 variants format (that is compatible with v4.3.)
• .fasta: FASTA file recording the consensus sequence calculated for
each reference contig.
Page 33
• .fastq: FASTQ file recording the consensus sequence calculated for
each reference contig, as well as per-base confidence scores.
Available Algorithms
At this time there are three algorithms available for variant calling:
plurality, poa and arrow.
• plurality is a simple and very fast procedure that merely tallies the
most frequent read base or bases found in alignment with each
reference base, and reports deviations from the reference as potential
variants. This is a very insensitive and flawed approach for PacBio
sequence data, and is prone to insertion and deletion errors.
• poa uses the partial order alignment algorithm to determine the
consensus sequence. It is a heuristic algorithm that approximates a
multiple sequence alignment by progressively aligning sequences to
an existing set of alignments.
• arrow uses the per-read SNR metric and the per-pulse pulsewidth
metric as part of its likelihood model.
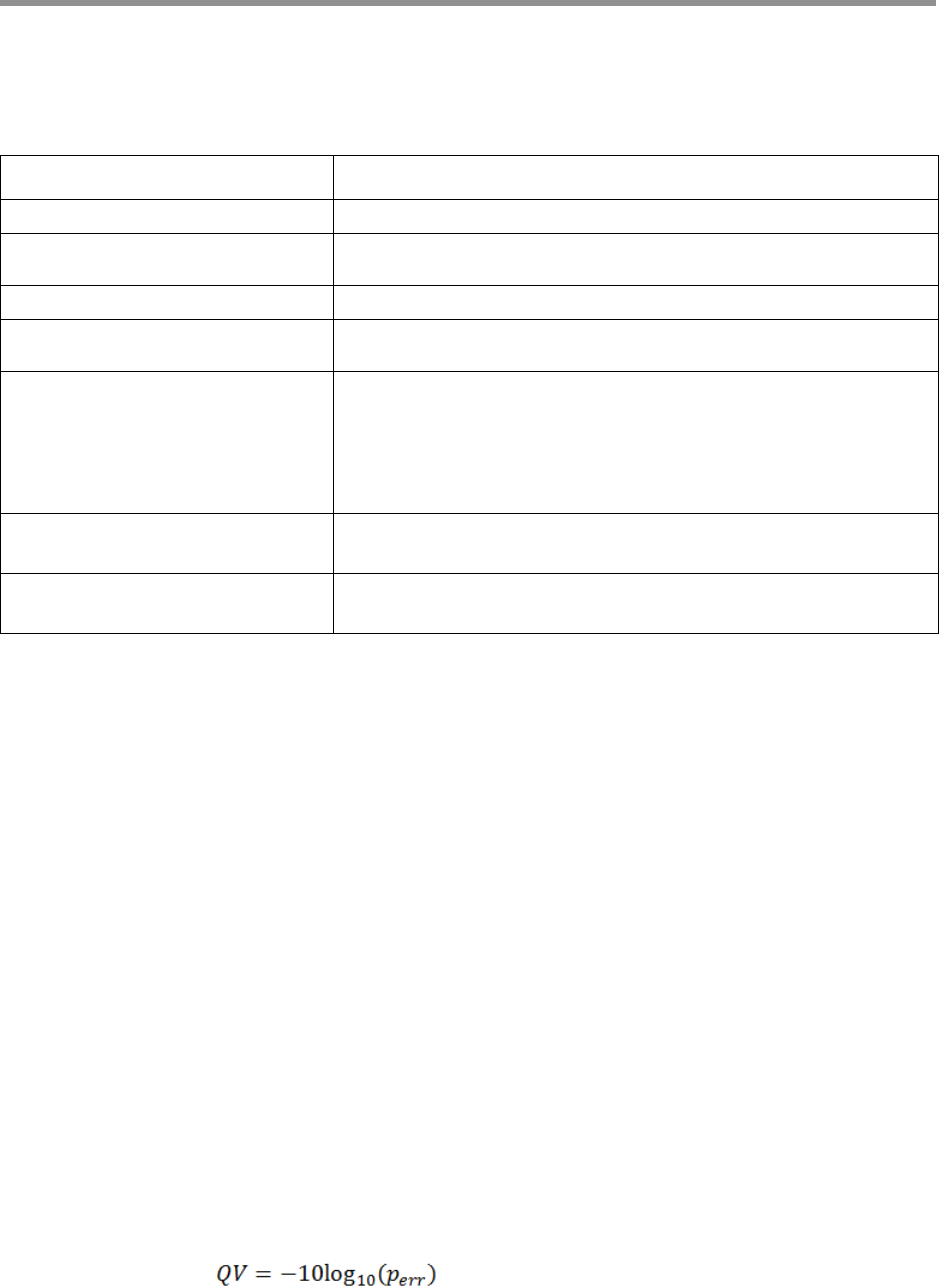
Confidence Values
The arrow and plurality algorithms make a confidence metric available
for every position of the consensus sequence. The confidence should be
interpreted as a phred-transformed posterior probability that the
consensus call is incorrect; such as:
Options Description
-j Specifies the number of worker processes to use.
--algorithm= Specifies the variant-calling algorithm to use; values are plurality and
arrow.
-r Specifies the FASTA reference file to use.
-o Specifies the output file format; values are .gff, .vcf, .fasta, and
.fastq.
--maskRadius When using the arrow algorithm, setting this option to a value N greater
than 0 causes gcpp to pass over the data a second time after masking out
regions of reads that have >70% errors in 2*N+1 bases. This setting has
little to no effect at low coverage, but for high-coverage datasets (>50X),
setting this parameter to 3 may improve final consensus accuracy. In rare
circumstances, such as misassembly or mapping to the wrong reference,
enabling this parameter may cause worse performance.
--minConfidence MINCONFIDENCE
-q MINCONFIDENCE
Specifies the minimum confidence for a variant call to be output to
variants.{gff,vcf} (Default = 40)
--minCoverage MINCOVERAGE
-x MINCOVERAGE
Specifies the minimum site coverage for variant calls and consensus to be
calculated for a site. (Default = 5)

Page 34
gcpp clips reported QV values at 93; larger values cannot be encoded in a
standard FASTQ file.
Chemistry Specificity
The --algorithm=arrow parameter is trained per-chemistry. arrow
identifies the sequencing chemistry used for each run by looking at
metadata contained in the input BAM data file. This behavior can be
overridden by a command-line option.
When multiple chemistries are represented in the reads in the input file,
the Arrow will model reads appropriately using the parameter set for its
chemistry, thus yielding optimal results.
ipdSummary
The ipdSummary tool detects DNA base-modifications from kinetic
signatures. It is part of the
kineticsTool package.
kineticsTool loads IPDs observed at each position in the genome,
compares those IPDs to value expected for unmodified DNA, and outputs
the result of this statistical test. The expected IPD value for unmodified
DNA can come from either an in-silico control or an amplified control. The
in-silico control is trained by Pacific Biosciences and shipped with the
package. It predicts the IPD using the local sequence context around the
current position. An amplified control Data Set is generated by sequencing
unmodified DNA with the same sequence as the test sample. An amplified
control sample is usually generated by whole-genome amplification of the
original sample.
Modification Detection
The basic mode of kineticsTool does an independent comparison of IPDs
at each position on the genome, for each strand, and outputs various
statistics to CSV and GFF files (after applying a significance filter).
Modifications Identification
kineticsTool also has a Modification Identification mode that can decode
multi-site IPD “fingerprints” into a reduced set of calls of specific
modifications. This feature has the following benefits:
• Different modifications occurring on the same base can be
distinguished; for example, 6mA and 4mC.
• The signal from one modification is combined into one statistic,
improving sensitivity, removing extra peaks, and correctly centering the
call.
Algorithm: Synthetic Control
Studies of the relationship between IPD and sequence context reveal that
most of the variation in mean IPD across a genome can be predicted from
a 12-base sequence context surrounding the active site of the DNA
polymerase. The bounds of the relevant context window correspond to the

Page 35
window of DNA in contact with the polymerase, as seen in DNA/
polymerase crystal structures. To simplify the process of finding DNA
modifications with PacBio data, the tool includes a pre-trained lookup table
mapping 12-mer DNA sequences to mean IPDs observed in C2 chemistry.
Algorithm: Filtering and Trimming
kineticsTool uses the Mapping QV generated by blasr and stored in the
cmp.h5 or BAM file (or AlignmentSet) to ignore reads that are not
confidently mapped. The default minimum Mapping QV required is 10,
implying that
blasr has 90% confidence that the read is correctly mapped.
Because of the range of read lengths inherent in PacBio data, this can be
changed using the
--mapQvThreshold option.
There are a few features of PacBio data that require special attention to
achieve good modification detection performance.
kineticsTool inspects
the alignment between the observed bases and the reference sequence
for an IPD measurement to be included in the analysis. The PacBio read
sequence must match the reference sequence for
k around the cognate
base. In the current module,
k=1. The IPD distribution at some locus can
be thought of as a mixture between the “normal” incorporation process
IPD, which is sensitive to the local sequence context and DNA
modifications, and a contaminating “pause” process IPD, which has a
much longer duration (mean > 10 times longer than normal), but happen
rarely (~1% of IPDs).
Note: Our current understanding is that pauses do not carry useful
information about the methylation state of the DNA; however a more
careful analysis may be warranted. Also note that modifications that
drastically increase the roughly 1% of observed IPDs are generated by
pause events. Capping observed IPDs at the global 99
th
percentile is
motivated by theory from robust hypothesis testing. Some sequence
contexts may have naturally longer IPDs; to avoid capping too much data
at those contexts, the cap threshold is adjusted per context as follows:
capThreshold = max(global99, 5*modelPrediction,
percentile(ipdObservations, 75))
Algorithm: Statistical Testing
We test the hypothesis that IPDs observed at a particular locus in the
sample have longer means than IPDs observed at the same locus in
unmodified DNA. If we have generated a Whole Genome Amplified Data
Set, which removes DNA modifications, we use a case-control, two-
sample t-test. This tool also provides a pre-calibrated “synthetic control”
model which predicts the unmodified IPD, given a 12-base sequence
context. In the synthetic control case we use a one-sample t-test, with an
adjustment to account for error in the synthetic control model.
Usage
To run using a BAM input, and output GFF and HDF5 files:

Page 36
ipdSummary aligned.bam --reference ref.fasta m6A,m4C --gff basemods.gff \
--csv_h5 kinetics.h5
To run using cmp.h5 input, perform methyl fraction calculation, and output
GFF and CSV files:
ipdSummary aligned.cmp.h5 --reference ref.fasta m6A,m4C --methylFraction \
--gff basemods.gff --csv kinetics.csv
Input Files
• A standard PacBio alignment file - either AlignmentSet XML, BAM, or
cmp.h5 - containing alignments and IPD information.
• Reference sequence used to perform alignments. This can be either a
FASTA file or a ReferenceSet XML.
Output Files
The tool provides results in a variety of formats suitable for in-depth
statistical analysis, quick reference, and consumption by visualization
tools such as SMRT View. Results are generally indexed by reference
position and reference strand. In all cases the strand value refers to the
strand carrying the modification in the DNA sample. Remember that the
kinetic effect of the modification is observed in read sequences aligning to
the opposite strand. So reads aligning to the positive strand carry
information about modification on the negative strand and vice versa, but
the strand containing the putative modification is always reported.
• modifications.gff: Compliant with the GFF Version 3 specification
(http://www.sequenceontology.org/gff3.shtml). Each template position/
strand pair whose probability value exceeds the probability value
threshold appears as a row. The template position is 1-based, per the
GFF specifications. The strand column refers to the strand carrying the
detected modification, which is the opposite strand from those used to
detect the modification. The GFF confidence column is a Phred-
transformed probability value of detection.
The auxiliary data column of the GFF file contains other statistics
which may be useful for downstream analysis or filtering. These
include the coverage level of the reads used to make the call, and +/-
20 bp sequence context surrounding the site.
Output Options Description
--gff FILENAME GFF format.
--csv FILENAME Comma-separated value format.
--csv_h5 FILENAME Compact binary-equivalent of .csv, in HDF5 format.
--bigwig FILENAME BigWig file format; mostly only useful for SMRT View.

Page 37
• modifications.csv: Contains one row for each (reference position,
strand) pair that appeared in the Data Set with coverage at least
x.
x defaults to 3, but is configurable with the --minCoverage option. The
reference position index is 1-based for compatibility with the GFF file in
the R environment. Note that this output type scales poorly and is not
recommended for large genomes; the HDF5 output should perform
much better in these cases.
Output Columns: In-Silico Control Mode
Output Columns: Case Control Mode
Column Description
refId Reference sequence ID of this observation.
tpl 1-based template position.
strand Native sample strand where kinetics were generated. 0 is the strand of the original
FASTA, 1 is opposite strand from FASTA.
base The cognate base at this position in the reference.
score Phred-transformed probability value that a kinetic deviation exists at this position.
tMean Capped mean of normalized IPDs observed at this position.
tErr Capped standard error of normalized IPDs observed at this position (standard
deviation/sqrt(coverage)).
modelPrediction Normalized mean IPD predicted by the synthetic control model for this sequence
context.
ipdRatio tMean/modelPrediction.
coverage Count of valid IPDs at this position.
frac Estimate of the fraction of molecules that carry the modification.
fracLow 2.5% confidence bound of the frac estimate.
fracUpp 97.5% confidence bound of the frac estimate.
Column Description
refId Reference sequence ID of this observation.
tpl 1-based template position.
strand Native sample strand where kinetics were generated. 0 is the strand of the
original FASTA, 1 is opposite strand from FASTA.
base The cognate base at this position in the reference.
score Phred-transformed probability value that a kinetic deviation exists at this
position.
caseMean Mean of normalized case IPDs observed at this position.
controlMean Mean of normalized control IPDs observed at this position.
caseStd Standard deviation of case IPDs observed at this position.
controlStd Standard deviation of control IPDs observed at this position.
ipdRatio tMean/modelPrediction.
testStatistic T-test statistic.

Page 38
isoseq3
The isoseq3 tool characterizes full-length transcripts. The analysis is
performed de novo, without a reference genome. The tool enables
analysis and functional characterization of transcript isoforms for
sequencing data generated on PacBio instruments.
Usage
isoseq3 <tool>
Typical workflow
1. Generate consensus sequences from raw subread data:
$ ccs movie.subreads.bam movie.ccs.bam --noPolish --minPasses 1
2. Remove primers and demultiplex:
$ cat primers.fasta
>primer_5p
AAGCAGTGGTATCAACGCAGAGTACATGGGG
>primer_3p
AAGCAGTGGTATCAACGCAGAGTAC
$ lima movie.ccs.bam primers.fasta demux.ccs.bam --isoseq --no-pbi
3. Remove noise from FL reads:
$ isoseq3 refine movie.fl.P5--P3.bam primers.fasta movie.flnc.bam
4. Cluster consensus sequences to generate unpolished transcripts:
$ isoseq3 cluster movie.flnc.bam unpolished.bam --verbose
5. Polish transcripts using subreads:
$ isoseq3 polish unpolished.bam movie.subreads.bam polished.bam
6. (Optional) Map transcripts to genome and collapse transcripts based
on genomic mapping:
$ pbmm2 align polished.bam reference.fasta aligned.sorted.bam --preset ISOSEQ --sort
$ isoseq3 collapse aligned.sorted.bam out.gff or
$ isoseq3 collapse aligned.sorted.bam movie.ccs.bam out.gff
cluster Tool: Cluster CCS reads and generate unpolished transcripts.
Usage
isoseq3 cluster [options] input output
coverage Mean of case and control coverage.
controlCoverage Count of valid control IPDs at this position.
caseCoverage Count of valid case IPDs at this position.
Column Description
Options Description
-h, --help Displays help information and exits.
--version Displays program version number and exits
Page 39
Example
isoseq3 cluster movie.consensusreadset.xml unpolished.bam
Custom BAM Tags
isoseq3 adds the following custom PacBio tags to the output BAM file:
• ib: Barcode summary - triplets delimited by semicolons; each triplet
contains two barcode indices and the ZMW counts; delimited by
comma. Example:
0,1,20;0,3,5
• im: ZMW names associated with this isoform.
• is: Number of ZMWs associated with this isoform.
• iz: Maximum number of subreads used for polishing.
• rq: Predicted accuracy for polished isoform.
polish Tool: Polish transcripts using subreads.
Usage
isoseq3 polish [options] input_1 input_2 output
Inputs/Outputs Description
input ccs.bam file or movie.consensusreadset.xml file.
output unpolished.bam file or unpolished.transcriptset.xml file.
Options Description
--require-polya Requires full-length reads to have a poly(A) tail and removes it.
--s1 Specifies the number of seeds for minimer-only clustering.
(Default = 1000)
--s2 Specifies the number of seeds for DP clustering. (Default = 1000)
--poa-cov Specifies the maximum number of CCS reads used for POA consensus.
(Default = 10)
--use-qvs Use CCS Quality Values; sets --poa-cov to 100.
--split-bam Splits BAM output files into a maximum of N files; 0 means no splitting.
(Default = 0)
--min-subreads-split Subread threshold for High-Quality/Low-Quality split; only works with
--use-qvs. (Default = 7)
--log-level Specifies the log level; values are [DEBUG, INFO, WARN, ERROR,
CRITICAL]. (Default = WARN)
-v,--verbose Uses verbose output.
-j,--num-threads Specifies the number of threads to use; 0 means autodetection.
(Default = 0)
--log-file Writes the log to a file. (Default = stdout)
--emit-tool-contract Outputs the tool contract to stdout. (Default = False)
--resolved-tool-contract Uses arguments from the resolved tool contract.

Page 40
Example
isoseq3 polish unpolished.bam movie.subreadset.xml polished.bam
summarize Tool: Create a .csv-format barcode overview from transcripts.
Usage
isoseq3 summarize [options] input output
Example
isoseq3 summarize polished.bam summary.csv
Inputs/Outputs Description
input_1 unpolished.bam file or unpolished.transcriptset.xml file.
input_2 movie.subreads.bam file or movie.subreadset.xml file.
output polished.bam file or polished.transcriptset.xml file.
Options Description
-r,--rq-cutoff Specifies the RQ cutoff for fastx output. (Default = 0.99)
-c,--coverage Specifies the maximum number of subreads used for polishing.
(Default = 60)
--log-level Specifies the log level; values are [DEBUG, INFO, WARN, ERROR,
CRITICAL]. (Default = WARN)
-v,--verbose Uses verbose output.
-j,--num-threads Specifies the number of threads to use; 0 means autodetection.
(Default = 0)
--log-file Writes the log to a file. (Default = stdout)
--emit-tool-contract Outputs the tool contract to stdout. (Default = False)
--resolved-tool-contract Uses arguments from the resolved tool contract.
Inputs/Outputs Description
input unpolished.bam file or unpolished.transcriptset.xml file.
output summary.csv file.
Options
--log-level Specifies the log level; values are [DEBUG, INFO, WARN, ERROR,
CRITICAL]. (Default = WARN)
-v,--verbose Uses verbose output.
--log-file Writes the log to file. (Default = stdout)
--emit-tool-contract Outputs the tool contract to stdout. (Default = False)
--resolved-tool-contract Uses arguments from the resolved tool contract.

Page 41
collapse Tool: Collapse transcripts based on genomic mapping.
Usage
isoseq3 collapse [options] <alignments.bam|xml> <ccs.bam|xml>
<out.fastq>
Examples:
isoseq3 collapse polished.aligned.sorted.bam out.gff
or
isoseq3 collapse polished.aligned.sorted.bam ccs.bam out.gff
juliet
juliet is a general-purpose minor variant caller that identifies and phases
minor single nucleotide substitution variants in complex populations. It
identifies codon-wise variants in coding regions, performs a reference-
guided de novo variant discovery, and annotates known drug-resistance
mutations. Insertion and deletion variants are currently ignored; support
will be added in a future version. There is no technical limitation with
respect to the target organism or gene.
The underlying model is a statistical test, the Bonferroni-corrected Fisher's
Exact test. It compares the number of observed mutated codons to the
number of expected mutations at a given position.
juliet uses JSON target configuration files to define different genes in
longer reference sequences, such as overlapping open reading frames in
HIV. These predefined configurations ease batch applications and allow
Inputs/Outputs Description
alignments Alignments mapping transcripts to the reference genome. (BAM or XML
file).
ccs.bam Optional input BAM file containing CCS sequences.
out.fastq Collapsed transcripts in FASTQ format.
Options Description
--min-aln-coverage Ignores alignments with less than the Minimum Query Coverage.
(Default = 0.95)
--min-aln-identity Ignores alignments with less than the Minimum Alignment Identity.
(Default = 0.50)
--max-fuzzy-junction Ignores mismatches or indels shorter than or equal to N. (Default = 5)
--version Displays program version number and exits.
--log-file Writes the log to file. (Default = stderr)
--log-level Specifies the log level; values are [DEBUG, INFO, WARN, ERROR,
CRITICAL]. (Default = WARN)
-j,--num-threads Specifies the number of threads to use; 0 means autodetection.
(Default = 0)

Page 42
immediate reproducibility. A target configuration may contain multiple
coding regions within one reference sequence and optional drug
resistance mutation positions.
Notes:
• The preinstalled target configurations are meant for a quick start. It is
the user's responsibility to ensure that the target configurations used
are correct and up-to-date.
• If the target configuration none was specified, the provided reference is
assumed to be in-frame.
Performance
At a coverage of 6,000 CCS reads with a predicted accuracy (RQ) of
≥
0.99, the false positive and false negative rates are below 1% and
0.001% (10
-5
), respectively.
Usage
$ juliet --config "HIV" data.align.bam patientZero.html
Required Description
input_file.bam Input aligned BAM file containing CCS records, which must be PacBio-
compliant, that is, cigar M is forbidden.
output_file.html Output report HTML file.
Configuration Description
--config,-c Path to the target configuration JSON file, predefined target configuration
tag, or the JSON string.
--mode-phasing,-p Phase variants and cluster haplotypes.
Restrictions Description
--region,-r Specifies the genomic region of interest; reads are clipped to that region.
Empty means all reads.
--drm-only,-k Only reports DRM positions specified in the target configuration. Can be
used to filter for drug-resistance mutations - only known variants from the
target configuration are called.
--min-perc,-m Specifies the minimum variant percentage to report.
Example: --min-perc 1 will only show variant calls with an observed
abundance of more than 1%. (Default = 0)
--max-perc,-n Specifies the maximum variant percentage to report.
Example: --max-perc 95 will only show variant calls with an observed
abundance of less than 95%. (Default = 100)
Page 43
Input Files
• BAM-format files containing CCS records. These must be PacBio-
compliant, that is,
cigar M is forbidden.
• Input CCS reads should have a minimal predicted accuracy of 0.99.
• Reads should be created with CCS2 using the --richQVs option.
Without the
--richQVs information, the number of false positive calls
might be higher, as
juliet is missing information to filter actual
heteroduplexes in the sample provided.
• juliet currently does not demultiplex barcoded data; you must
provide one BAM file per barcode.
Output Files
A JSON and/or HTML file:
$ juliet data.align.bam patientZero.html
$ juliet data.align.bam patientZero.json
$ juliet data.align.bam patientZero.html patientZero.json
The HTML file includes the same content as the JSON file, but in more
human-readable format. The HTML file contains four sections:
1. Input Data
Summarizes the data provided, the exact call for juliet, and juliet
version for traceability purposes.
2. Target Config
Summarizes details of the provided target configuration for traceability.
This includes the configuration version, reference name and length, and
Chemistry Override (Specify
both)
Description
--sub,-s Specifies the substitution rate. Use to override the learned rate.
(Default = 0)
--del,-d Specifies the deletion rate. Use to override the learned rate. (Default = 0)
Options Description
--help, -h Displays help information and exits.
--verbose, -v Sets the verbosity level.
--version Displays program version number and exits.
--debug Returns all amino acids, irrespective of their relevance.
--emit-tool-contract Emits the tool contract.
--resolved-tool-contract Uses arguments from the resolved tool contract.
--mode-phasing,-p Phases variants and cluster haplotypes.

Page 44
annotated genes. Each gene name (in bold) is followed by the reference
start, end positions, and possibly known drug resistance mutations.
3. Variant Discovery
For each gene/open reading frame, there is one overview table.
Each row represents a variant position.
• Each variant position consists of the reference codon, reference amino
acid, relative amino acid position in the gene, mutated codon,
percentage, mutated amino acid, coverage, and possible affected
drugs.
• Clicking the row displays counts of the multiple-sequence alignment
counts of the -3 to +3 context positions.

Page 45
4. Drug Summaries
Summarizes the variants grouped by annotated drug mutations:
Predefined Target Configuration
juliet ships with one predefined target configuration, for HIV. Following
is the command syntax for running that predefined target configuration:
$ juliet --config "HIV" data.align.bam patientZero.html
Page 46
• Note: For the predefined configuration HIV, please use the HIV HXB2
complete genome for alignment.
Customized Target Configuration
To define your own target configuration, create a JSON file. The root child
genes contains a list of coding regions, with begin and end, the name of
the gene, and a list of drug resistant mutations. Each DRM consists of its
name and the positions it targets. The
drms field is optional. If provided,
the
referenceSequence is used to call mutations, otherwise it will be
tested against the major codon. All indices are with respect to the provided
alignment space, 1-based, begin-inclusive and end-exclusive
[).
Target Configuration Example 1- A customized json target configuration
file named
my_customized_hiv.json:
{
"genes": [
{
"begin": 2550,
"drms": [
{
"name": "fancy drug",
"positions": [ "M41L" ]
}
],
"end": 2700,
"name": "Reverse Transcriptase"
}
],
"referenceName": "my seq",
"referenceSequence": "TGGAAGGGCT...",

Page 47
"version": "Free text to version your config files"
"databaseVersion": "DrugDB version x.y.z (last updated YYYY-MM-DD)"
}
Run with a customized target configuration using the --config option:
$ juliet --config my_customized_hiv.json data.align.bam patientZero.html
Valid Formats for DRMs/positions
103 Only the reference position.
M130 Reference amino acid and reference position.
M103L Reference aa, reference position, mutated aa.
M103LKA Reference aa, reference position, list of possible mutated aas.
103L Reference position and mutated aa.
103LG Reference position and list mutated aas.
Missing amino acids are processed as wildcard (*).
Example:
{ "name": "ATV/r", "positions": [ "V32I", "L33", "46IL",
"I54VTALM", "V82ATFS", "84" ] }
Target Configuration Example 2 - BCR-ABL:
For BCR-ABL, using the ABL1 gene with the following reference
NM_005157.5 (https://www.ncbi.nlm.nih.gov/nuccore/NM_005157.5) a
typical target configuration looks like this:
{
"genes": [
{
"name": "ABL1",
"begin": 193,
"end": 3585,
"drms": [
{
"name": "imatinib",
"positions": [
"T315AI","Y253H","E255KV","V299L","F317AICLV","F359CIV" ]
},
{
"name": "dasatinib",
"positions": [ "T315AI","V299L","F317AICLV" ]
},
{
"name": "nilotinib",
"positions": [ "T315AI","Y253H","E255KV","F359CIV" ]
},
{
"name": "bosutinib",
"positions": [ "T315AI" ]
}
]
}
],
"referenceName": "NM_005157.5",
"referenceSequence": "TTAACAGGCGCGTCCC..."

Page 48
No Target Configuration
If no target configuration is specified, either make sure that the sequence
is in-frame, or specify the region of interest to mark the correct reading
frame, so that amino acids are correctly translated. The output is labeled
with
unknown as the gene name:
$ juliet data.align.bam patientZero.html
Phasing
The default mode is to call amino-acid/codon variants independently.
Using the
--mode-phasing option, variant calls from distinct haplotypes
are clustered and visualized in the HTML output.
• The row-wise variant calls are "transposed" onto per-column
haplotypes. Each haplotype has an ID:
[A-Z]{1}[a-z]?.
• For each variant, colored boxes in this row mark haplotypes that
contain this variant.
• Colored boxes per haplotype/column indicate variants that co-occur.
Wild type (no variant) is represented by plain dark gray. A color palette
helps to distinguish between columns.
• The JSON variant positions has an additional haplotype_hit boolean
array with the length equal to the number of haplotypes. Each entry
indicates if that variant is present in the haplotype. A haplotype block
under the root of the JSON file contains counts and read names. The
order of those haplotypes matches the order of all
haplotype_hit
arrays.

Page 49
There are two types of tooltips in the haplotype section of the table.
The first tooltip is for the Haplotypes % and shows the number of reads
that count towards (A) Actually reported haplotypes, (B) Haplotypes that
have less than 10 reads and are not being reported, and (C) Haplotypes
that are not suitable for phasing. Those first three categories are mutually
exclusive and their sum is the total number of reads going into
juliet. For
(C), the three different marginals provide insights into the sample quality;
as they are marginals, they are not exclusive and can overlap. The
following image shows a sample with bad PCR conditions:
The second type of tooltip is for each haplotype percentage and shows the
number of reads contributing to this haplotype:
laa
Long Amplicon Analysis (LAA) finds phased consensus sequences from a
pooled set of (possibly polyploid) amplicons sequenced with Pacific
Biosciences’ SMRT technology. Sometimes referred to as LAA2, the
executable
laa is a complete rewrite of the AmpliconAnalysis module
from the
ConsensusTools package included with earlier versions of SMRT
Analysis, which performed a similar function in the Quiver framework.
laa is a computational and memory-intensive software tool that builds
upon the Arrow framework for generating high-quality consensus
sequences. It is generally preferable to run
laa using the SMRT Link
interface for efficient distribution across a compute cluster. However, it is
occasionally useful to run
laa from the command-line to identify optimal
parameter settings or to diagnose a problem.
Run Modes
AmpliconAnalysis is a general solution for the analysis of PCR products
generated with SMRT sequencing, and it can be run in multiple
configurations depending on the design of the experiment.

Page 50
1. Pooled Polyploid Amplicons: The default mode assumes that the
data contains a single complex mixture of amplicons, which may come
from different genes and may have multiple alleles.
2. Barcoded Polyploid Amplicons: If passed a file of barcoding results,
AmpliconAnalysis will instead separate the data by barcode and run
the above process on each subset.
3. Barcoded Simple Amplicons: Another common use case is to
generate consensus sequences for a large number of simple ampli-
cons, such as for synthetic construct validation or high-throughput
screening.
Input Files
laa only accepts PacBio-compatible BAM files or Data Set XML files as
input.
In addition, the underlying files themselves now contain barcode
information. This document assumes that you already have a barcoded
PacBio BAM file containing the data to be analyzed.
Output Files
laa produces two sets of FASTQ files containing a sequence for each
phased template sequence in each coarse cluster, and for each barcode.
• amplicon_analysis.fastq: Contains all of the high-quality non-
artifactual sequences found.
• amplicon_analysis_chimeras_noise.fastq: Contains sequences
thought to be some form of PCR or sequencing artifact.
Note: A sequence is defined as an artifact if, in the summary CSV file,
the value of either the
IsDuplicate, NoiseSequence or IsChimera
column is
True.
• amplicon_analysis_summary.csv: Contains summary information
about each read. Empty fields and values of
-1 represent inapplicable
columns, while fields with
1 represent True and 0 represents False.
Contains the following fields:
– BarcodeName: Name of the barcode the reads came from. This is set
to
0 for non-barcode runs.
– FastaName: Sequence ID or header string.
– CoarseCluster: Number of the coarse cluster the sequence came
from.
– Phase: Number of the phase of the sequence in the coarse cluster.
– TotalCoverage: Total number of subreads mapped to this
sequence. This may be capped using the
numPhasingReads option.
– SequenceLength: Length of this consensus sequence.
– ConsensusConverged: 1 if a final consensus was reached within the
allotted iterations;
0 if otherwise. 0 may indicate problems with the
underlying sample or data.

Page 51
– PredictedAccuracy: Predicted accuracy of the consensus
sequence, calculated by multiplying together the QVs generated by
Arrow.
– NoiseSequence: 1 if the sequence has a low-quality consensus,
corresponding to a predicted accuracy less than 95% indicating a
probable PCR artifact;
0 if otherwise.
– IsDuplicate: 1 if the sequence is a duplicate of another with more
coverage;
0 if otherwise.
– DuplicateOf: If IsDuplicate is 1, contains the name of the other
sequence; otherwise empty.
– IsChimera: 1 if the sequence is tagged as a chimeric by the
UCHIME-like chimera labeler;
0 if otherwise.
– ChimeraScore: UCHIME-like score for sequences tested as
possible chimeras.
– ParentSequenceA: If chimeric, the name of the consensus thought
to be the source of the left half.
– ParentSequenceB: If chimeric, the name of the consensus thought
to be the source of the right half.
– CrossoverPosition: Position in the chimeric sequence where the
junction between the parent sequences is thought to have occurred.
• amplicon_analysis_subreads.X.csv: Contains mapping
probabilities for each subread used to call the consensus sequences
generated. A separate file is written for each barcode pair, where
X is
replaced with the name of that pair. Contains the following fields:
– SubreadId: The name of a particular subread used in the current
run.
– <A Consensus Sequence Name>: The mapping probability for the
subread listed in SubreadId to the particular consensus sequence
named.
Usage
laa [options] INPUT
Options Description
-h, --help Displays help information and exits.
--verbose, -v Sets the verbosity level.
--version Displays program version number and exits.
--log level Sets the logging level. (Default = INFO)
--rngSeed RNG seed, modulates reservoir filtering of reads. (Default = 42)
--generateBamIndex Generates PacBio indicies (*.pbi) for BAM files that don't have them.
--ignoreBamIndex Ignores PacBio indicies (*.pbi) for BAM files if they exist.
-M,--modelPath Specifies the path to a model file or directory containing model files.
-m,--modelSpec Specifies the name of chemistry or model to use, overriding the default
selection.
--numThreads,-n Specifies the number of threads to use; 0 means autodetection.
(Default = 0)

Page 52
--takeN Reports only the top N consensus sequences for each barcode. To
disable, use a number less than 1. (Default = 0)
-t,--trimEnds Trims N bases from each end of each consensus. (Default = 0)
--minPredictedAccuracy Specifies the minimum predicted consensus accuracy below which a
consensus is treated as noise. (Default = 0.949999988079071)
--chimeraScoreThreshold Specifies the minimum score to consider a sequence chimeric.
(Default = 1)
--ChimeraFilter Activates the chimera filter and separate chimeric consensus outputs.
--noChimeraFilter Deactivates the chimera filter and outputs all consensus.
--logFile Output file to write logging information to.
--resultFile Output file name for high-quality results.
(Default = amplicon_analysis.fastq)
--junkFile Output file name for low-quality or chimeric results.
(Default = amplicon_analysis_chimeras_noise.fastq)
--reportFile Output file name for the summary report.
(Default = amplicon_analysis_summary.csv)
--inputReportFile Output file name for the output estimates of input PCR quality, based on
subread mappings. (Default = amplicon_analysis_input.csv)
--subreadsReportPrefix Prefix for the output subreads report.
(Default = amplicon_analysis_subreads)
-b,--barcodes Specifies the FASTA file name of the barcode sequences used, which
overwrites any barcode names in the Data Set. Note: This is used only
to find barcode names.
--minBarcodeScore Specifies the minimum average barcode score required for subreads.
(Default = 0)
--fullLength Filters input reads by presence of both flanking barcodes.
--doBc Specifies a comma-separated list of barcode pairs to analyse. This can be
by name ("lbc1--lbc1") or by Index ("0--0").
--ignoreBc Disables barcode filtering so that all data be treated as one sample.
-l,--minLength Specifies the minimum length of input reads to use. (Default = 3000)
-L,--maxLength Specifies the maximum length of input reads to use. To disable, set to 0.
(Default = 0)
-s,--minReadScore Specifies the minimum read score of input reads to use. (Default = 0.75)
--minSnr Specifies the minimum SNR of input reads to use. (Default = 3.75)
--whitelist Specifies a file of ReadIds, in either Text or FASTA format, to allow from
the input file. (Default = NONE)
-r,--maxReads Specifies the maximum number of input reads, per barcode, to use in
analysis. (Default = 2000)
-c,--maxClusteringReads Specifies the maximum number of input reads to use in the initial
clustering step. (Default = 500)
--fullLengthPreference Prefers full-length subreads when clustering.
--fullLengthClustering Uses only full-length subreads when clustering.
--clusterInflation Markov clustering inflation parameter. (Default = 2)
Options Description

Page 53
Algorithm Description
laa proceeds in six main phases: Data filtering, coarse clustering,
waterfall clustering, fine phasing, consensus polishing, and post-
processing.
• Data filtering is used to separate out sequences by their barcode
calls, if present, so that only reads long enough to meaningfully
contribute to phasing are used.
• The Coarse and Waterfall Clustering steps are used to find and
separate reads coming from different amplicons.
• The reads from each cluster are then put through the phasing step,
which recursively separates full-length haplotypes using a variant of
the Arrow model. Those haplotypes are then polished within the
Arrow framework to achieve a high-quality consensus sequence.
--clusterLoopWeight Markov clustering loop weight parameter.
(Default = 0.00100000004749745)
--skipRate Skips some high-scoring alignments to disperse the cluster more.
(Default = 0.0)
--minClusterSize Specifies the minimum number of reads supporting a cluster before it is
reported. (Default = 20)
--doCluster Only analyzes one specified cluster. (Default = -1)
--Clustering Enables coarse clustering.
--noClustering Disables coarse clustering.
-i,--ignoreEnds When splitting, ignores N bases at the end. This prevents excessive
splitting caused by degenerate primers. (Default = 0)
--maxPhasingReads Specifies the maximum number of reads to use for phasing/consensus.
(Default = 500)
--minQScore Specifies the minimum score to require of mutations used for phasing.
(Default = 20)
--minPrevalence Specifies the minimum prevalence to require of mutations used for
phasing. (Default = 0.0900000035762787)
--minSplitReads Specifies the minimum number of reads favoring the minor phase required
to split a haplotype. (Default = 20)
--minSplitFraction Specifies the minimum fraction of reads favoring the minor phase required
to split a haplotype. (Default = 0.100000001490116)
--minSplitScore Specifies the global likelyhood improvement required to split a haplotype.
(Default = 500)
--minZScore Specifies the minimum Z Score to allow before adding a read to a
haplotype. (Default = -10)
--Phasing Enables the fine phasing step.
--noPhasing Disables the fine phasing step.
--emit-tool-contract Emits the tool contract.
--resolved-tool-contract Uses arguments from the resolved tool contract.
Options Description

Page 54
• Finally, a post-processing step attempts to identify and remove
spurious consensus sequences and sequences representing PCR
artifacts.
Data Filtering
In this first step, we separate sequences by barcode and then apply a
series of user-selected quality filters to speed up down-stream processing
and improve result quality. Filters are used primarily to remove short
subreads (which may not be long enough to phase variants of interest)
and subreads with low barcode scores (representing reads for whom the
barcode call is uncertain and may be incorrect). A “Whitelist” option is also
available so that users can specify the exact list of subreads or ZMWs to
use.
Coarse Clustering and Waterfall Clustering
The coarse clustering step groups the number of subreads (set by the
maxClusteringReads option) that originate from different amplicons into
different clusters. It works by detecting subread-to-subread similarities,
building a graph of the results, and then clustering nodes (subreads) using
the Markov Clustering algorithm (http://micans.org/mcl/). The Markov
clustering step is needed to remove spurious similarities caused by
chimeric reads that can arise from PCR errors or doubly-loaded ZMWs, or
just by chance due to sequencing error.
Next, if the number of subreads specified with the maxReads option is
greater than the number used in coarse clustering, any remaining
subreads are aligned to a rough consensus of each cluster and added to
the cluster with the greatest similarity. This “waterfall” step allows for a
larger number of reads to be used much more quickly than if all subreads
had to be clustered using the normal coarse clustering process.
At the end of clustering, subreads in each cluster are then sorted for
downstream analysis using the PageRank algorithm (Page, Lawrence, et
al. “The PageRank citation ranking: Bringing order to the web.” (1999)).
This ensures that the most representative reads of the cluster are used
first in the generation of consensus sequences.
Phasing/Consensus
The reads assigned to each cluster are loaded into the Arrow framework,
and an initial consensus of all reads is found. SNP differences between
subreads and the initial consensus are scored with the Arrow model, and
combinations of high-scoring SNPs are tested for their ability to segregate
the reads into multiple haplotypes. If sufficient evidence of a second
haplotype is found, the template sequence is “split” into two copies, one
with the SNPs applied to the template and one without. This process is
repeated recursively so long as new haplotypes with sufficient scores can
be found with at least some minimum level of coverage.

Page 55
Post-Processing Filters
laa implements a post-processing step to flag likely PCR artifacts in the
set of phased output sequences. First, consensus sequences that are
identical duplicates of other consensus sequences in the results are
removed. Next, those with unusually low predicted accuracy are flagged
as being probable sequencing artifacts and removed. PacBio implemented
a filter for consensus sequences from PCR crossover events, which on
average make up ~5 to 20% of products generated by PCR amplifications
>3 kb in length.
For artifacts of PCR crossover events, or “chimeras”, PacBio implemented
a variant of the UCHIME algorithm (Edgar, Robert C., et al. “UCHIME
improves sensitivity and speed of chimera detection.” Bioinformatics
27.16(2011): 2194-2200). The consensus sequences are sorted in order of
decreasing read coverage, and the first two sequences are accepted as
non-chimeric since they have no possible parent sequences with greater
coverage. The remaining sequences are evaluated in descending order,
with each test sequence aligned to all non-chimeric sequences so far
processed. Crossovers between pairs of non-chimeric sequences are
checked to see if they would yield a sequence very similar to the test
sequence. If one is found with a sufficient score, the test sequence is
marked as chimeric. If not, the test sequence is added to the list of non-
chimeric sequences.
motifMaker
The motifMaker tool identifies motifs associated with DNA modifications
in prokaryotic genomes. Modified DNA in prokaryotes commonly arises
from restriction-modification systems that methylate a specific base in a
specific sequence motif. The canonical example is the m6A methylation of
adenine in GATC contexts in E. coli. Prokaryotes may have a very large
number of active restriction-modification systems present, leading to a
complicated mixture of sequence motifs.
PacBio SMRT sequencing is sensitive to the presence of methylated DNA
at single base resolution, via shifts in the polymerase kinetics observed in
the real-time sequencing traces. For more background on modification
detection, see
http://nar.oxfordjournals.org/content/early/2011/12/07/nar.gkr1146.full.
Algorithm
Existing motif-finding algorithms such as MEME-chip and YMF are sub-
optimal for this case for the following reasons:
• They search for a single motif, rather than attempting to identify a
complicated mixture of motifs.
• They generally don't accept the notion of aligned motifs - the input to
the tools is a window into the reference sequence which can contain
the motif at any offset, rather than a single center position that is
available with kinetic modification detection.

Page 56
• Implementations generally either use a Markov model of the reference
(MEME-chip), or do exact counting on the reference, but place
restrictions on the size and complexity of the motifs that can be
discovered.
Following is a rough overview of the algorithm used by motifMaker:
Define a motif as a set of tuples: (position relative to methylation, required
base). Positions not listed in the motif are implicitly degenerate. Given a
list of modification detections and a genome sequence, define the
following objective function on motifs:
Motif score(motif) = (# of detections matching motif) / (# of genome sites matching
motif) * (Sum of log-pvalue of detections matching motif) = (fraction methylated) * (sum
of log-pvalues of matches)
Then, search (close to exhaustively) through the space of all possible
motifs, progressively testing longer motifs using a branch-and-bound
search. The “fraction methylated” term must be less than 1, so the
maximum achievable score of a child node is the sum of scores of
modification hits in the current node, allowing pruning of all search paths
whose maximum achievable score is less than the best score discovered
so far.
Usage
Run the find command, and pass the reference FASTA and the
modifications.gff (.gz) file output by the PacBio modification detection
workflow.
The reprocess subcommand annotates the GFF file with motif
information for better genome browsing.
MotifMaker [options] [command] [command options]
find Command: Run motif-finding.
find [options]
reprocess Command: Update a modifications.gff file with motif
information based on new Modification QV thresholds.
Options Description
-h, --help Displays help information and exits.
* -f, --fasta Reference FASTA file.
* -g, --gff Modifications.gff or .gff.gz file.
-m, --minScore Specifies the minimum Qmod score to use in motif finding.
(Default = 40.0)
* -o, --output Outputs motifs.csv file.
-x, --xml Outputs motifs XML file.
Page 57
reprocess [options]
Output Files
Using the find command:
• Output CSV file: This file has the same format as the standard "Fields
included in motif_summary.csv" described in the Methylome Analysis
White Paper (https://github.com/PacificBiosciences/Bioinformatics-
Training/wiki/Methylome-Analysis-Technical-Note).
Using the reprocess command:
• Output GFF file: The format of the output file is the same as the input
file, and is described in the Methylome Analysis White Paper under
"Fields included in the modifications.gff file" (https://github.com/
PacificBiosciences/Bioinformatics-Training/wiki/Methylome-Analysis-
Technical-Note).
pbalign
The pbalign tool aligns PacBio reads to reference sequences; filters
aligned reads according to user-specified filtering criteria; and converts the
output to PacBio BAM, SAM, or PacBio Data Set format.
Input Files
The pbalign tool distinguishes input and output file formats by file
extensions. The tool supports the following input formats:
• BAM: .bam
• Data Set: .subreadset.xml or .consensusreadset.xml
• FASTA: .fa or .fasta
• File-Of-File-Names: .fofn
The input reference sequences can be in a FASTA file or a reference Data
Set.
Options Description
-c, --csv Raw modifications.csv file.
* -f, --fasta Reference FASTA file.
* -g, --gff Modifications.gff or .gff.gz file.
-m, --minFraction Specifies that only motifs above this methylated fraction are used.
(Default = 0.75)
-m, --motifs Motifs.csv file.
* -o, --output Reprocessed modifications.gff file.

Page 58
Output Files
The tool supports the following output formats:
• BAM: .bam
• Data Set: .xml
• SAM: .sam
Usage
pbalign [-h] [--verbose] [--version] [--profile] [--debug]
[--regionTable REGIONTABLE] [--configFile CONFIGFILE]
[--algorithm {blasr,bowtie}] [--maxHits MAXHITS]
[--minAnchorSize MINANCHORSIZE]
[--maxMatch MAXMATCH]
[--useccs {useccs,useccsall,useccsdenovo}]
[--noSplitSubreads] [--nproc NPROC]
[--algorithmOptions ALGORITHMOPTIONS]
[--maxDivergence MAXDIVERGENCE] [--minAccuracy MINACCURACY]
[--minLength MINLENGTH]
[--scoreFunction {alignerscore,editdist,blasrscore}]
[--scoreCutoff SCORECUTOFF]
[--concordant]
[--hitPolicy {randombest,allbest,random,all}] [--forQuiver]
[--seed SEED] [--tmpDir TMPDIR]
inputFileName referencePath outputFileName
Required Description
inputFileName The input file of PacBio reads. Can be a BAM, Data Set, FASTA file, or a
fofn (File-Of-File-Names).
referencePath Either a reference FASTA file or a PacBio reference Data Set file.
outputFileName The output .bam, .xml or .sam file.
Options Description
-h, --help Displays help information and exits.
--verbose, -v Sets the verbosity level.
--version Displays program version number and exits.
--profile Prints runtime profile at exit.
--debug Runs within a debugger session.
--configFile Specifies a set of user-defined argument values.
--algorithm Selects an algorithm from blasr or bowtie. (Default = blasr)
--maxHits Specifies the maximum number of matches of each read to the reference
sequence to evaluate. (Default = 10)
--minAnchorSize Specifies the length of the read that must match against the reference
sequence. (Default = 12)
--maxMatch Stops extending an anchor between the read and the reference sequence
when its length reaches this value. Bypasses the blasr maxMatch option.
(Default = 30)
Page 59
Examples
Basic usage:
$ pbalign tests/data/example/read.bam \
tests/data/example/ref.fasta \
tests/data/example/example.bam
Basic usage with optional arguments:
$ pbalign --maxHits 10 --hitPolicy all \
tests/data/example_read.fasta \
tests/data/example_ref.fasta \
--noSplitSubreads Does not split reads into subreads even if subread regions are available.
(Default = False)
--concordant Maps subreads of a ZMW to the same genomic location.
(Default = False)
--nproc NPROC Specifies the number of threads. (Default = 8)
--algorithmOptions Passes alignment options through. Note: By default, blasr places gap
inconsistently when aligning a sequence and its reverse complement
sequence. It is preferable to place gap consistently to call a consensus
sequence from multiple alignments or call single nucleotide variants
(SNPs), as the output alignments will make it easier for variant callers to
call variants. To do so, specify
--algorithmOptions=’ –-placeGapConsistently’.
--maxDivergence Specifies the maximum allowed percentage divergence of a read from the
reference sequence. (Default = 30)
--minAccuracy Specifies the minimum percentage accuracy of alignments to evaluate.
(Default = 70)
--minLength Specifies the minimum aligned read length of alignments to evaluate.
(Default = 50)
--scoreFunction Specifies a score function for evaluating alignments.
• alignerscore: Aligner's score in the SAM tag as.
• editdist: Edit distance between read and reference.
• blasrscore: The blasr default score function.
(Default = alignerscore)
--scoreCutoff Specifies the worst score to output an alignment.
--hitPolicy Specifies a policy for how to treat multiple hits.
• random: Selects a random hit.
• all: Selects all hits.
• allbest: Selects all the best score hits.
• randombest: Selects a random hit from all best alignment score hits.
• leftmost: Reports an alignment which has the best alignment score
and has the smallest mapping coordinates in any reference.
(Default = randombest)
--seed Initializes the random number generator with a non-zero integer. 0 means
that current system time is used. (Default = 1)
--tmpDir Specifies a directory for saving temporary files. (Default = /scratch)
Options Description

Page 60
example.sam
Advanced usage - To import predefined options from a configuration file:
$ pbalign --configFile=tests/data/1.config \
tests/data/example/read.fasta \
tests/data/example/ref.fasta \
example.sam
Advanced usage - To pass options through to the Aligner:
$ pbalign --algorithmOptions='-nCandidates 10 -sdpTupleSize 12 --placeGapConsistently'\
tests/data/example/read.fasta \
tests/data/example/ref.fasta \
example.sam \
Advanced usage - To use pbalign as a library using the Python API:
$ python
>>> from pbalign.pbalignrunner import PBAlignRunner
>>> # Specify arguments in a list.
>>> args = ['--maxHits', '20', 'tests/data/example/read.fasta',\
... 'tests/data/example/ref.fasta', 'example.sam']
>>> # Create a PBAlignRunner object.
>>> a = PBAlignRunner(args)
>>> # Execute.
>>> exitCode = a.start()
>>> # Show all files used.
>>> print a.fileNames
pbcromwell
The pbcromwell tool is Pacific Biosciences’ wrapper for the cromwell
scientific workflow engine used to power SMRT Link. pbcromwell
includes advanced utilities for interacting directly with a Cromwell server.
pbcromwell is designed primarily for running workflows distributed and
supported by PacBio, but it is written to handle any valid WDL source
(version 1.0), and is very flexible in how it takes input. PacBio workflows
are expected to be found in the directory defined by the
SMRT_PIPELINE_BUNDLE_DIR environment variable, which is automatically
defined by the SMRT Link distribution.
Note that pbcromwell does not interact with SMRT Link services; to run
a
Cromwell workflow as a SMRT Link job, please use pbservice. (For
details, see
“pbservice” on page 71.)
Note: Interaction with the Cromwell server is primarily intended for
developers and power users.
Usage
pbcromwell {run,show-workflows,show-workflow-details,configure,submit,get-
job,abort,metadata,show-running,wait}

Page 61
Enter pbcromwell {command} -h for a command's options.
Examples:
Show available PacBio-developed workflows:
$ pbcromwell --quiet show-workflows
Show details for a PacBio workflow:
$ pbcromwell --quiet show-workflow-details pb_ccs
Generate cromwell.conf with HPC settings:
$ pbcromwell configure --default-backend SGE --output-file cromwell.conf
Launch a PacBio workflow:
$ pbcromwell run pb_ccs -e /path/to/movie.subreadset.xml --nproc 8 --config /full/
path/to/cromwell.conf
pbcromwell run Command: Run a Cromwell workflow. Multiple input
modes are supported, including a pbsmrtpipe-like set of arguments
(for PacBio workflows only), and JSON files already in the native
Cromwell format.
Usage:
pbcromwell run [-h] [--output-dir OUTPUT_DIR] [--overwrite] [-i INPUTS]
[-e ENTRY_POINTS] [-n NPROC] [-c MAX_NCHUNKS]
[--target-size TARGET_SIZE] [--queue QUEUE] [-o OPTIONS]
[-t TASK_OPTIONS] [-b BACKEND] [-r MAX_RETRIES]
[--tmp-dir TMP_DIR] [--config CONFIG] [--dry-run]
workflow
Options Description
-h, --help Displays help information and exits.
--version Displays program version number and exits.
--log-file LOG_FILE Writes the log to file. (Default = None, writes to stdout.)
--log-level=INFO Specifies the log level; values are [DEBUG, INFO, WARNING, ERROR,
CRITICAL.] (Default = INFO)
--debug Alias for setting the log level to DEBUG. (Default = False)
--quiet Alias for setting the log level to CRITICAL to suppress output.
(Default = False)
--verbose, -v Sets the verbosity level. (Default = None)
Options Description
--output-dir OUTPUT_DIR Output directory for Cromwell output. (Default = cromwell_out)

Page 62
All PacBio workflows have similar requirements to the pbsmrtpipe
pipelines in previous SMRT Link versions:
1. One or more PacBio dataset XML entry points, usually a SubreadSet
or ConsensusReadSet (
--entry-point <FILE>.)
2. Any number of workflow-specific task options (--task-option
<OPTION>
.)
3. Engine options independent of the workflow, such as number of pro-
cessors per task (--nproc), or compute backend (--backend).
Output is directed to a new directory: --output-dir, which defaults to
cromwell_out. This includes final inputs for the Cromwell CLI, and
subdirectories for logs (workflow and task outputs), links to output files,
and the Cromwell execution itself, which has a complex nested directory
structure. Detailed information about the workflow execution can be found
in the file
metadata.json, in the native Cromwell format.
Note that output file links do not include the individual resource files of
datasets and reports (BAM files, index files, plot PNGs, and so on.) Follow
the symbolic links to their real path (for example using
readlink -f) to
find report plots.
--overwrite Overwrites the output directory, if it exists. (Default = False)
-i INPUTS, --inputs INPUTS Cromwell inputs and settings as JSON files. (Default = None)
-e ENTRY_POINTS, --entry
ENTRY_POINTS
Entry point Data Set; may be repeated for workflows that take more than
one input Data Set. Note that all PacBio workflows require at least one
such entry point.
-n NPROC, --nproc NPROC Number of processors per task. (Default = 1)
-c MAX_NCHUNKS, --max-nchunks
MAX_NCHUNKS
Maximum number of chunks per task. (Default = None)
--target-size TARGET_SIZE Target chunk size. (Default = None)
--queue QUEUE Cluster queue to use. (Default = None)
-o OPTIONS, --options OPTIONS Additional Cromwell engine options, as a JSON file. (Default = None)
-t TASK_OPTIONS, --task-
option TASK_OPTIONS
Workflow- or task-level option as key=value strings, specific to the
application. May be specified multiple times for multiple options.
(Default = [])
-b BACKEND, --backend BACKEND Backend to use for running tasks. (Default = None)
-r MAX_RETRIES, --maxRetries
MAX_RETRIES
Maximum number of times to retry a failing task. (Default = 1)
--tmp-dir TMP_DIR Optional temporary directory for Cromwell tasks, which must exist on all
compute hosts. (Default = None)
--config CONFIG Java configuration file for running Cromwell. (Default = None)
--dry-run Don't execute Cromwell, just write final inputs and then exit.
(Default = True)
workflow Workflow ID (such as pb_ccs or cromwell.workflows.pb_ccs for
PacBio workflows only) or q path to a Workflow Description Language
(WDL) source file.
Options Description

Page 63
For further information about Cromwell, consult the official documentation
at https://cromwell.readthedocs.io.
Workflow Examples:
Run the CCS workflow:
$ pbcromwell run pb_ccs -e <SUBREADS> --nproc 8 --config /full/path/to/cromwell.conf
Run the Iso-Seq workflow, including mapping to a reference, and execute
on SGE:
$ pbcromwell run pb_isoseq3 -e <SUBREADS> -e <PRIMERS> -e <REFERENCE> --nproc 8 --
config /full/path/to/cromwell.conf
Run a user-defined workflow:
$ pbcromwell run my_workflow.wdl -i inputs.json -o options.json --config /full/path/to/
cromwell.conf
Set up input files but exit before starting Cromwell:
$ pbcromwell run pb_ccs -e <SUBREADS> --nproc 8 --dry-run
Print details about the named PacBio workflow, including input files and
task options. Note: The prefix
cromwell.workflows. is optional.
$ pbcromwell show-workflow-details pb_ccs
$ pbcromwell show-workflow-details cromwell.workflows.pb_ccs
pbdagcon
The pbdagcon tool implements DAGCon (Directed Acyclic Graph
Consensus), which is a sequence consensus algorithm based on using
directed acyclic graphs to encode multiple sequence alignments.
pbdagcon uses the alignment information from blasr to align sequence
reads to a "backbone" sequence. Based on the underlying alignment
directed acyclic graph (DAG), it uses the new information from the reads to
find the discrepancies between the reads and the "backbone" sequences.
A dynamic programming process is then applied to the DAG to find the
optimum sequence of bases as the consensus. The new consensus can
be used as a new backbone sequence to iteratively improve the
consensus quality.
While the code is developed for processing Pacific Biosciences raw
sequence data, the algorithm can be used for general consensus
purposes. Currently, it only takes FASTA input. For shorter read
sequences, one might need to adjust the
blasr alignment parameters to
get the alignment string properly.
Note: This code is not an official Pacific Biosciences software release.

Page 64
Examples
To generate consensus from blasr alignments:
This is the most basic use case to generate a consensus from a set of
alignments by directly using the
pbdagcon executable.
At the most basic level, pbdagcon takes information from blasr
alignments sorted by target and generates FASTA-formatted corrected
target sequences. The alignments from
blasr can be formatted with either
-m 4 or -m 5. For -m 4 format, the alignments must be run through a
format adapter (
m4topre.py) to generate suitable input to pbdagcon.
The following example shows the simplest way to generate a consensus
for one target using
blasr -m 5 alignments as input:
blasr queries.fasta target.fasta -bestn 1 -m 5 -out mapped.m5
pbdagcon mapped.m5 > consensus.fasta
To generate corrected reads from daligner alignments:
Support for generating consensus from daligner output exists as a new
executable:
dazcon. Note that dazcon is sensitive to the version of
daligner used and may fail if using inputs generated by versions other
than what is referenced in the submodules.
dazcon -ox -j 4 -s subreads.db -a subreads.las > corrected.fasta
To correct PacBio reads using HGAP:
This example shows how PacBio reads are corrected in PacBio's
"Hierarchical Genome Assembly Process" (HGAP) workflow. HGAP uses
blasr -m 4 output.
This example makes use of the filterm4.py and m4topre.py scripts:
# First filter the m4 file to help remove chimeras:
filterm4.py mapped.m4 > mapped.m4.filt
# Next run the m4 adapter script, generating 'pre-alignments':
m4topre.py mapped.m4.filt mapped.m4.filt reads.fasta 24 > mapped.pre
# Finally, correct using pbdagcon, typically using multiple consensus threads:
pbdagcon -j 4 -a mapped.pre > corrected.fasta
pbindex
The pbindex tool creates an index file that enables random access to
PacBio-specific data in BAM files.

Page 65
Usage
pbindex <input>
Input File
• *.bam file containing PacBio data.
Output File
• *.pbi index file, with the same prefix as the input file name.
pbmm2
The pbmm2 tool aligns native PacBio data, outputs PacBio BAM files, and
is a SMRT minimap2 wrapper for PacBio data.
Note: pbmm2 is the official replacement for blasr and pbalign.
pbmm2 supports native PacBio input and output, provides sets of
recommended parameters, generates sorted output on-the-fly, and post-
processes alignments. Sorted output can be used directly for polishing
using GenomicConsensus, if BAM has been used as input to pbmm2.
Benchmarks show that pbmm2 runs faster than blasr and outperforms it
in mapped concordance and number of mapped bases.
pbmm2 adds the following SAM tags to each aligned record:
• mc, stores mapped concordance percentage between 0.0 and 100.0.
• rm, is set to 1 if alignment has been manipulated by repeated matches
trimming.
Usage
pbmm2 <tool>
index Command: Indexes references and stores them as .mmi files.
Indexing is optional, but recommended if you use the same refer-
ence with the same --preset multiple times.
Usage:
pbmm2 index [options] <ref.fa|xml> <out.mmi>
Options Description
-h, --help Displays help information and exits.
--version Displays program version number and exits.
Options Description
-h, --help Displays help information and exits.
--version Displays program version number and exits.

Page 66
Input File
• *.fasta, *.fa file containing reference contigs or
*.referenceset.xml.
Output File
• out.mmi (minimap2 index file.)
Notes:
• You can use existing minimap2 .mmi files with pbmm2 align.
• If you use an index file, you cannot override parameters -k, -w, nor -u
in
pbmm2 align.
• The minimap2 parameter -H (homopolymer-compressed k-mer) is
always on for SUBREAD and UNROLLED presets, and can be
disabled using
-u.
align Command: Aligns PacBio reads to reference sequences. The
output argument is optional; if not provided, the BAM output is
streamed to stdout.
Usage:
pbmm2 align [options] <ref.fa|xml|mmi> <in.bam|xml|fa|fq> [out.aligned.bam|xml]
Input Files
• *.fasta file containing reference contigs, or *.referenceset.xml, or
*.mmi index file.
• *.bam, *.subreadset.xml, *.consensusreadset.xml,
*.transcriptset.xml, *.fasta, *.fa, *.fastq, or *.fastq file
containing PacBio data.
Output Files
• *.bam aligned reads in BAM format.
• *.alignmentset, *.consensusalignmentset.xml, or
*.transcriptalignmentset.xml if XML output was chosen.
The following Data Set Input/output combinations are allowed:
Options Description
--preset Specifies the alignment mode:
• "SUBREAD" -k 19 -w 10
• "CCS" -k 19 -w 10 -u
• "ISOSEQ" -k 15 -w 5 -u
• "UNROLLED" -k 15 -w 15
(Default = SUBREAD)
-k Specifies the k-mer size, which cannot be larger than 28. (Default = -1)
-w Specifies the Minimizer window size. (Default = -1)
-u,--no-kmer-compression Disables homopolymer-compressed k-mer. (Compression is on by default
for the SUBREAD and UNROLLED presets.)
Page 67
SubreadSet > AlignmentSet
pbmm2 align hg38.referenceset.xml movie.subreadset.xml hg38.movie.alignmentset.xml
ConsensusReadSet > ConsensusAlignmentSet
pbmm2 align hg38.referenceset.xml movie.consensusreadset.xml
hg38.movie.consensusalignmentset.xml --preset CCS
TranscriptSet > TranscriptAlignmentSet
pbmm2 align hg38.referenceset.xml movie.transcriptset.xml
hg38.movie.transcriptalignmentset.xml --preset ISOSEQ
FASTA/Q input
In addition to native PacBio BAM input, reads can also be provided in
FASTA and FASTQ formats.
Attention: The resulting output BAM file cannot be used as input into
GenomicConsensus!
With FASTA/Q input, the --rg option sets the read group. Example:
pbmm2 align hg38.fasta movie.Q20.fastq hg38.movie.bam --preset CCS --rg
'@RG\tID:myid\tSM:mysample'
All three reference file formats .fasta, .referenceset.xml, and .mmi
can be combined with FASTA/Q input.
Options Description
-h, --help Displays help information and exits.
--chunk-size Processes N records per chunk. (Default = 100)
--sort Generates a sorted BAM file.
-m,--sort-memory Specifies the memory per thread for sorting. (Default = 768M)
-j,--alignment-threads Specifies the number of threads used for alignment. 0 means
autodetection. (Default = 0)
-J,--sort-threads Specifies the number of threads used for sorting. 0 means 25% of -j, with
a maximum of 8. (Default = 0)
--sample Specifies the sample name for all read groups. Defaults, in order of
precedence: A) SM field in the input read group B) Biosample name
C) Well sample name D) "UnnamedSample".
--rg Specifies the read group header line such as '@RG\tID:xyz\tSM:abc'.
Only for FASTA/Q inputs.
-c,--min-concordance-perc Specifies the minimum alignment concordance, in percent. (Default = 70)
-l,--min-length Specifies the minimum mapped read length, in base pairs. (Default = 50)
-N,--best-n Specifies the output at maximum N alignments for each read. 0 means no
maximum. (Default = 0)
--strip Removes all kinetic and extra QV tags. The output cannot be polished.

Page 68
Examples:
Generate an index file for reference and reuse it to align reads:
--split-by-sample Specifies one output BAM file per sample.
--no-bai Omits BAI index file generation for sorted output.
--unmapped Specifies that unmapped records be included in the output.
--median-filter Picks one read per ZMW of median length.
--zmw Processes ZMW Reads; subreadset.xml input is required. This
activates the UNROLLED preset.
--hqregion Processes the HQ region of each ZMW; subreadset.xml input is
required. This activates the UNROLLED preset.
Parameter Set Options and
Overrides
Description
--preset Specifies the alignment mode:
• "SUBREAD" -k 19 -w 10 -o 5 -O 56 -e 4 -E 1 -A 2 -B 5
-z 400 -Z 50 -r 2000 -L 0.5
• "CCS" -k 19 -w 10 -u -o 5 -O 56 -e 4 -E 1 -A 2 -B 5 -
z 400 -Z 50 -r 2000 -L 0.5
• "ISOSEQ" -k 15 -w 5 -u -o 2 -O 32 -e 1 -E 0 -A 1 -B 2
-z 200 -Z 100 -C 5 -r 200000 -G 200000 -L 0.5
• "UNROLLED" -k 15 -w 15 -o 2 -O 32 -e 1 -E 0 -A 1 -B 2
-z 200 -Z 100 -r 2000 -L 0.5
(Default = SUBREAD)
-k Specifies the k-mer size, which cannot be no larger than 28. (Default = -1)
-w Specifies the Minimizer window size. (Default = -1)
-u,--no-kmer-compression Disables homopolymer-compressed k-mer. (Compression is on by default
for the SUBREAD and UNROLLED presets.)
-A Specifies the matching score. (Default = -1)
-B Specifies the mismatch penalty. (Default = -1)
-z Specifies the Z-drop score. (Default = -1)
-Z Specifies the Z-drop inversion score. (Default = -1)
-r Specifies the bandwidth used in chaining and DP-based alignment.
(Default = -1)
-o,--gap-open-1 Specifies the gap open penalty 1. (Default = -1)
-O,--gap-open-2 Specifies the gap open penalty 2. (Default = -1)
-e,--gap-extend-1 Specifies the gap extension penalty 1. (Default = -1)
-E,--gap-extend-2 Specifies the gap extension penalty 2. (Default = -1)
-L,--lj-min-ratio Specifies the long join flank ratio. (Default = -1)
-G Specifies the maximum intron length; this changes -r. (Default = -1)
-C Specifies the cost for a non-canonical GT-AG splicing. (Default = -1)
--no-splice-flank Specifies that you do not prefer splicing flanks GT-AG.
Options Description

Page 69
$ pbmm2 index ref.fasta ref.mmi
$ pbmm2 align ref.mmi movie.subreads.bam ref.movie.bam
Align reads and sort on-the-fly, with 4 alignment and 2 sort threads:
$ pbmm2 align ref.fasta movie.subreads.bam ref.movie.bam --sort -j 4 -J 2
Align reads, sort on-the-fly, and create a PBI:
$ pbmm2 align ref.fasta movie.subreadset.xml ref.movie.alignmentset.xml --sort
Omit the output file and stream the BAM output to stdout:
$ pbmm2 align hg38.mmi movie1.subreadset.xml | samtools sort > hg38.movie1.sorted.bam
Align the CCS fastq input and sort the output:
$ pbmm2 align ref.fasta movie.Q20.fastq ref.movie.bam --preset CCS --sort --rg
'@RG\tID:myid\tSM:mysample'
Alignment Parallelization
The number of alignment threads can be specified using the -j,--
alignment-threads
option. If not specified, the maximum number of
threads will be used, minus one thread for BAM I/O and minus the number
of threads specified for sorting.
Sorting
Sorted output can be generated using the --sort option.
• By default, 25% of threads specified with the -j option (Maximum = 8)
are used for sorting.
• To override the default percentage, the -J,--sort-threads option
defines the explicit number of threads used for on-the-fly sorting. The
memory allocated per sort thread is defined using the
-m,--sort-
memory
option, accepting suffixes M,G.
Benchmarks on human data show that 4 sort threads are recommended,
but that no more than 8 threads can be effectively leveraged, even with 70
cores used for alignment. We recommend that you provide more memory
to each of a few sort threads to avoid disk I/O pressure, rather than
providing less memory to each of many sort threads.
What are parameter sets and how can I override them?
Per default, pbmm2 uses recommended parameter sets to simplify the
multitudes of possible combinations. Please see the available parameter
sets in the option table shown earlier.
What other special parameters are used implicitly?
To achieve alignment behavior similar to blasr, we implicitly use the
following
minimap2 parameters:

Page 70
• Soft clipping with -Y.
• Long cigars for tag CG with -L.
• X/= cigars instead of M with --eqx.
• No overlapping query intervals with repeated matches trimming.
• No secondary alignments are produced using the --secondary=no
option.
What is repeated matches trimming?
A repeated match occurs when the same query interval is shared between
a primary and supplementary alignment. This can happen for
translocations, where breakends share the same flanking sequence:
And sometimes, when a LINE gets inserted, the flanks are duplicated,
leading to complicated alignments, where we see a split read sharing a
duplication. The inserted region itself, mapping to a random other LINE in
the reference genome, may also share sequence similarity to the flanks:
To get the best alignments, minimap2 decides that two alignments may
use up to 50% (default) of the same query bases. This does not work for
PacBio, as
pbmm2 is a blasr replacement and requires that a single base
may never be aligned twice.
Minimap2 offers a feature to enforce a query
interval overlap to 0%. If a query interval gets used in two alignments, one
or both get flagged as secondary and get filtered. This leads to yield loss,
and more importantly, missing SVs in the alignment.
Papers (such as this) present dynamic programming approaches to find
the optimal split to uniquely map query intervals, while maximizing
alignment scores. We don't have per base alignment scores available,
thus our approach is much simpler. We align the read, find overlapping
query intervals, determine one alignment to be maximal reference-
spanning, then trim all others. By trimming,
pbmm2 rewrites the cigar and
the reference coordinates on-the-fly. This allows us to increase the
number of mapped bases, which slightly reduces mapped concordance,
but boosts SV recall rate.
How can I set the sample name?
You can override the sample name (SM field in the RG tag) for all read
groups using the
--sample option. If not provided, sample names derive

Page 71
from the Data Set input using the following order of precedence: A) SM field
in the input read group B) Biosample name C) Well sample name
D) UnnamedSample. If the input is a BAM file and the --sample option was
not used, the
SM field will be populated with UnnamedSample.
Can I split output by sample name?
Yes, the --split-by-sample option generates one output BAM file per
sample name, with the sample name as the file name prefix, if there is
more than one aligned sample name.
Can I remove all those extra per-base and per-pulse tags?
Yes, the --strip option removes the following extraneous tags if the input
is BAM:
dq, dt, ip, iq, mq, pa, pc, pd, pe, pg, pm, pq, pt,
pv, pw, px, sf, sq, st
. Note that the resulting output BAM file cannot
be used as input into
GenomicConsensus.
Where are the unmapped reads?
Per default, unmapped reads are omitted. You can add them to the output
BAM file using the
--unmapped option.
Can I output at maximum the N best alignments per read?
Use the option -N, --best-n. If set to 0, (the default), maximum filtering
is disabled.
Is there a way to only align one subread per ZMW?
Using the --median-filter option, only the subread closest to the
median subread length per ZMW is aligned. Preferably, full-length
subreads flanked by adapters are chosen.
pbservice
The pbservice tool performs a variety of useful tasks within SMRT Link.
• To get help for pbservice, use pbservice -h.
• To get help for a specific pbservice command, use
pbservice <command> -h.
Note: Starting in SMRT Link v6.0.0, pbservice now requires
authentication when run from a remote host, using the same credentials
used to log in to the SMRT Link GUI. (This also routes all requests through
HTTPS port 8243, so the services port is not required if authentication is
used.) Access to services running on
localhost will continue to work
without authentication.
All pbservice commands include the following optional parameters:
Options Description
--host=http://localhost Specifies the server host. Override the default with the environmental
variable PB_SERVICE_HOST.

Page 72
status Command: Use to get system status.
pbservice status [-h] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO}
[--debug] [--quiet]
import-dataset Command: Import Local Data Set XML. The file location
must be accessible from the host where the services are running; often on
a shared file system.
pbservice import-dataset [-h] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO]
[--debug] [--quiet]
xml_or_dir
import-fasta Command: Import a FASTA file and convert to a
ReferenceSet file. The file location must be accessible from the host
where the services are running; often on a shared file system.
pbservice import-fasta [-h] --name NAME --organism ORGANISM --ploidy
PLOIDY [--block] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO]
[--debug] [--quiet]
--port=8070 Specifies the server port. Override the default with the environmental
variable PB_SERVICE_PORT.
--log-file LOG_FILE Writes the log to file. (Default = None, writes to stdout.)
--log-level=INFO Specifies the log level; values are [DEBUG, INFO, WARNING, ERROR,
CRITICAL.] (Default = INFO)
--debug=False Alias for setting the log level to DEBUG. (Default = False)
--quiet=False Alias for setting the log level to CRITICAL to suppress output.
(Default = False)
--user USERNAME Specifies the user to authenticate as; this is required if the target host is
anything other than localhost.
--ask-pass Prompts the user to enter a password.
--password PASSWORD Supplies the password directly. This exposes the password in the shell
history (or log files), so this option is not recommended unless you are
using a limited account without Unix login access.
Options Description
Required Description
xml_or_dir Specifies a directory or XML file for the Data Set.

Page 73
fasta_path
run-analysis Command: Run a secondary analysis pipeline using an
analysis.json file.
pbservice run-analysis [-h] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO]
[--debug] [--quiet] [--block]
json_path
emit-analysis-template Command: Output an analysis.json
template to
stdout that can be run using the run-analysis command.
pbservice emit-analysis-template [-h] [--log-file LOG_FILE]
[--log-level INFO]
[--debug] [--quiet]
get-job Command: Get a job summary by Job Id.
pbservice get-job [-h] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO]
[--debug] [--quiet]
job_id
get-jobs Command: Get job summaries by Job Id.
pbservice get-jobs [-h] [-m MAX_ITEMS] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO]
Required Description
fasta_path Path to the FASTA file to import.
Options Description
--name Specifies the name of the ReferenceSet to convert the FASTA file to.
--organism Specifies the name of the organism.
--ploidy Ploidy.
--block=False Blocks during importing process.
Required Description
json_path Path to the analysis.json file.
Options Description
--block=False Blocks during importing process.
Required Description
job_id Job id or UUID.

Page 74
[--debug] [--quiet]
get-dataset Command: Get a Data Set summary by Data Set Id or
UUID.
pbservice get-dataset [-h] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO]
[--debug] [--quiet]
id_or_uuid
get-datasets Command: Get a Data Set list summary by Data Set type.
pbservice get-datasets [-h] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO]
[--debug] [--quiet] [-m MAX_ITEMS]
[-t DATASET_TYPE]
delete-dataset Command: Delete a specified Data Set.
Note: This is a "soft" delete - the database record is tagged as inactive so
it won't display in any lists, but the files will not be removed.
pbservice delete-dataset [-h] [--host HOST] [--port PORT]
[--log-file LOG_FILE]
[--log-level INFO]
[--debug] [--quiet]
[ID]
Examples
To obtain system status, the Data Set summary, and the job summary:
pbservice status --host smrtlink-release --port 9091
To import a Data Set XML:
pbservice import-dataset --host smrtlink-release --port 9091 \
path/to/subreadset.xml
Options Description
-m=25, --max-items=25 Specifies the maximum number of jobs to get.
Required Description
id_or_uuid Data Set Id or UUID.
Required Description
-t=subreads, --dataset-
type=subreads
Specifies the type of Data Set to retrieve: subreads, alignments,
references, barcodes.
Required Description
ID A valid Data Set ID, either UUID or integer ID, for the Data Set to delete.

Page 75
To obtain a job summary using the Job Id:
pbservice get-job --host smrtlink-release --port 9091 \
--log-level CRITICAL 1
To obtain Data Sets by using the Data Set Type subreads:
pbservice get-datasets --host smrtlink-alpha --port 8081 \
--quiet --max-items 1 -t subreads
To obtain Data Sets by using the Data Set Type alignments:
pbservice get-datasets --host smrtlink-alpha --port 8081 \
--quiet --max-items 1 -t alignments
To obtain Data Sets by using the Data Set Type references:
pbservice get-datasets --host smrtlink-alpha --port 8081 \
--quiet --max-items 1 -t references
To obtain Data Sets by using the Data Set Type barcodes:
pbservice get-datasets --host smrtlink-alpha --port 8081 \
--quiet --max-items 1 -t barcodes
To obtain Data Sets by using the Data Set UUID:
pbservice get-dataset --host smrtlink-alpha --port 8081 \
--quiet 43156b3a-3974-4ddb-2548-bb0ec95270ee
pbsv
pbsv is a structural variant caller for PacBio reads. It identifies structural
variants and large indels (Default: ≥20 bp) in a sample or set of samples
relative to a reference.
pbsv identifies the following types of variants:
Insertions, deletions, duplications, copy number variants, inversions, and
translocations.
pbsv takes as input read alignments (BAM) and a reference genome
(FASTA); it outputs structural variant calls (VCF).
Usage:
pbsv [-h] [--version] [--quiet] [--verbose]
{discover,call}...
Options Description
-h, --help Displays help information and exits.
--version Displays program version number and exits.
--log-file Logs to a file, instead of stdout.
--log-level Specifies the log level; values are [TRACE,DEBUG,INFO, WARN,
FATAL.] (Default = WARN)
discover Finds structural variant signatures in read alignments (BAM to SVSIG).

Page 76
pbsv discover
This command finds structural variant (SV) signatures in read alignments.
The input read alignments must be sorted by chromosome position.
Alignments are typically generated with
pbmm2. The output SVSIG file
contains SV signatures.
Usage:
pbsv discover [options] <ref.in.bam|xml> <ref.out.svsig.gz>
pbsv call
This command calls structural variants from SV signatures and assigns
genotypes.
The input SVSIG file is generated using pbsv discover. The output is
structural variants in VCF format.
call Calls structural variants from SV signatures and assign genotypes (SVSIG
to VCF).
Options Description
Required Description
ref.in.bam|xml Coordinate-sorted aligned reads in which to identify SV signatures.
ref.out.svsig.gz Structural variant signatures output.
Options Description
-h, --help Displays help information and exits.
-s,--sample Overrides sample name tag from BAM read group.
-q,--min-mapq Ignores alignments with mapping quality < N. (Default = 20)
-m,--min-ref-span Ignores alignments with reference length < N bp. (Default = 100)
-w,--downsample-window-length Specifies a window in which to limit coverage, in base pairs.
(Default = 10K)
-a,--downsample-max-
alignments
Considers up to N alignments in a window; 0 means disabled.
(Default = 20)
-r,--region Limits discovery to this reference region: CHR|CHR:START-END.
-l,--min-svsig-length Ignores SV signatures with length < N bp. (Default = 7)
-b,--tandem-repeats Specifies tandem repeat intervals for indel clustering, as an input BED file.
-k,--max-skip-split Ignores alignment pairs separated by > N bp of a read or reference.
(Default = 100)

Page 77
Usage:
pbsv call [options] <ref.fa|xml> <ref.in.svsig.gz|fofn>
<ref.out.vcf>
Required Description
ref.fa|xml Reference FASTA file or ReferenceSet XML file against which to call
variants.
ref.in.svsig.gz|fofn SV signatures from one or more samples. This can be either an SV
signature SVSIG file generated by pbsv discover, or a FOFN of SVSIG
files.
ref.out.vcf Variant call format (VCF) output file.
Options Description
-h, --help Displays help information and exits.
-j,--num-threads Specifies the number of threads to use, 0 means autodetection.
(Default = 0)
-z,--chunk-length Processes in chunks of N reference bp. (Default = “1M”)
-t,--types Calls these SV types: "DEL", "INS", "INV", "DUP", "BND", "CNV".
(Default = “DEL,INS,INV,DUP,BND,CNV”)
-m,--min-sv-length Ignores variants with length < N bp. (Default = 20)
--min-cnv-length Ignore CNVs with length < N bp. (Default = 1K)
--max-inversion-gap Does not link inverted alignments with > N bp gap or overlap with flanking
alignments. (Default = 1K)
--cluster-max-length-perc-
diff
Does not cluster signatures with difference in length > P%. (Default = 25)
--cluster-max-ref-pos-diff Does not cluster signatures > N bp apart in the reference. (Default = 200)
--cluster-min-basepair-perc-
id
Does not cluster signatures with base pair identity < P%. (Default = 10)
-x,--max-consensus-coverage Limits to N reads for variant consensus. (Default = 20)
-s,--poa-scores Scores POA alignment with triplet match,mismatch,gap.
(Default = “1,-2,-2“)
--min-realign-length Considers segments with > N length for realignment. (Default = 100)
-A, --call-min-reads-all-
samples
Ignores calls supported by < N reads total across samples. (Default = 2)
-O, --call-min-reads-one-
sample
Ignores calls supported by < N reads in every sample. (Default = 2)
-S, --call-min-reads-per-
strand-all-samples
Ignore calls supported by < N reads per strand total across samples.
(Default = 1)
-P, --call-min-read-perc-one-
sample
Ignores calls supported by < P% of reads in every sample. (Default = 20)
--ccs CCS optimized parameters: -A 1 -O 1 -S 0 -P 10.
--gt-min-reads Specifies the minimum supporting reads to assign a sample a non-
reference genotype. (Default = 1)
--annotations Annotates variants by comparing with sequences in FASTA.
(Default annotations are ALU, L1, and SVA.)

Page 78
Following is a typical SV analysis workflow starting from subreads:
1. Align PacBio reads to a reference genome, per movie:
Subreads BAM Input:
pbmm2 align ref.fa movie1.subreads.bam ref.movie1.bam --sort --median-filter --sample
sample1
CCS BAM Input:
pbmm2 align ref.fa movie1.ccs.bam ref.movie1.bam --sort --preset CCS --sample sample1
CCS FASTQ Input:
pbmm2 align ref.fa movie1.Q20.fastq ref.movie1.bam --sort --preset CCS --sample sample1
--rg '@RG\tID:movie1'
2. Discover the signatures of structural variation, per movie or per
sample:
pbsv discover ref.movie1.bam ref.sample1.svsig.gz
pbsv discover ref.movie2.bam ref.sample2.svsig.gz
3. Call structural variants and assign genotypes (all samples); for CCS
input append
--ccs:
pbsv call ref.fa ref.sample1.svsig.gz ref.sample2.svsig.gz
ref.var.vcf
Launching a Multi-Sample pbsv Analysis - Requirements
1. Merge multiple Bio Sample SMRT Cells to one Data Set with the Bio
Samples specified.
– Each SMRT Cell must have exactly one Bio Sample name - multiple
Bio Sample names cannot be assigned to one SMRT Cell.
– Multiple SMRT Cells can have the same Bio Sample name.
– All of the inputs need to already have the appropriate Bio Sample
records in their
CollectionMetadata. If they don't, they are treated
as a single sample.
2. Create a ReferenceSet from a FASTA file.
– The ReferenceSet is often already generated and registered in
SMRT Link.
– If the ReferenceSet doesn’t exist, use the dataset create
command to create one:
--annotation-min-perc-sim Annotates variant if sequence similarity > P%. (Default = 60)
--min-N-in-gap Considers ≥ N consecutive "N" bp as a reference gap. (Default = 50)
--filter-near-reference-gap Flags variants < N bp from a gap as "NearReferenceGap".
(Default = 1000)
--filter-near-contig-end Flags variants < N bp from a contig end as “NearContigEnd”.
(Default = 1K)
Options Description

Page 79
dataset create --type ReferenceSet --name reference_name reference.fasta
Launching a Multi-Sample Analysis
# Set subreads and ref FASTA
sample1=sample1.subreadset.xml sample2=sample2.subreadset.xml
ref=reference.fasta
pbmm2 align ${ref} ${sample1} sample1.bam --sort --median-filter --sample Sample1
pbmm2 align ${ref} ${sample2} sample2.bam --sort --median-filter --sample Sample2
samtools index sample1.bam
samtools index sample2.bam
pbindex sample1.bam
pbindex sample2.bam
pbsv discover sample1.bam sample1.svsig.gz
pbsv discover sample2.bam sample2.svsig.gz
pbsv call ${ref} sample1.svsig.gz sample2.svsig.gz out.vcf
out.vcf: A pbsv VCF output file, where columns starting from column 10
represent structural variants of Sample 1 and Sample 2:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1 Sample2
chr01 222737 pbsv.INS.1 T TTGGTGTTTGTTGTTTTGTTTT . PASS
SVTYPE=INS;END=222737;SVLEN=21;SVANN=TANDEM GT:AD:DP 0/1:6,4:10 0/1:6,5:11
pbvalidate
The pbvalidate tool validates that files produced by PacBio software are
compliant with Pacific Biosciences’ own internal specifications.
Input Files
pbvalidate supports the following input formats:
• BAM
• FASTA
• Data Set XML
See http://pacbiofileformats.readthedocs.org/en/5.1/ for further information
about each format's requirements.
Usage
pbvalidate [-h] [--version] [--log-file LOG_FILE]
[--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL} | --debug | --quiet | -v]
[-c] [--quick] [--max MAX_ERRORS]
[--max-records MAX_RECORDS]
[--type
{BAM,Fasta,AlignmentSet,ConsensusSet,ConsensusAlignmentSet,SubreadSet,BarcodeSet,Conti
gSet,ReferenceSet,HdfSubreadSet}]
[--index] [--strict] [-x XUNIT_OUT] [--unaligned]
[--unmapped] [--aligned] [--mapped]
[--contents {SUBREAD,CCS}] [--reference REFERENCE]
file
Required Description
file Input BAM, FASTA, or Data Set XML file to validate.

Page 80
Examples
To validate a BAM file:
$ pbvalidate in.subreads.bam
To validate a FASTA file:
$ pbvalidate in.fasta
Options Description
-h, --help Displays help information and exits.
--version Displays program version number and exits.
--log-file LOG_FILE Writes the log to file. Default (None) will write to stdout.
--log-level Specifies the log level; values are [DEBUG, INFO, WARNING, ERROR,
CRITICAL.] (Default = CRITICAL)
--debug=False Alias for setting the log level to DEBUG. (Default = False)
--quiet Alias for setting the log level to CRITICAL to suppress output.
(Default = False)
--verbose, -v Sets the verbosity level. (Default = None)
--quick Limits validation to the first 100 records (plus file header); equivalent to
--max-records=100. (Default = False)
--max MAX_ERRORS Exits after MAX_ERRORS were recorded.
(Default = None; checks the entire file.)
--max-records MAX_RECORDS Exits after MAX_RECORDS were inspected.
(Default = None; checks the entire file.)
--type Uses the specified file type instead of guessing.
[BAM,Fasta,AlignmentSet,ConsensusSet,ConsensusAlignmen
tSet,SubreadSet,BarcodeSet,ContigSet,ReferenceSet,
HdfSubreadSet] (Default = None)
--index Requires index files:.fai or .pbi. (Default = False)
--strict Turns on additional validation, primarily for Data Set XML.
(Default = False)
BAM Options Description
--unaligned Specifies that the file should contain only unmapped alignments.
(Default = None, no requirement.)
--unmapped Alias for --unaligned. (Default = None)
--aligned Specifies that the file should contain only mapped alignments.
(Default = None, no requirement.)
--mapped Alias for --aligned. (Default = None)
--contents Enforces the read type: [SUBREAD, CCS] (Default = None)
--reference REFERENCE Specifies the path to an optional reference FASTA file, used for additional
validation of mapped BAM records. (Default = None)

Page 81
To validate a Data Set XML file:
$ pbvalidate in.subreadset.xml
To validate a BAM file and its index file (.pbi):
$ pbvalidate --index in.subreads.bam
To validate a BAM file and exit after 10 errors are detected:
$ pbvalidate --max 10 in.subreads.bam
To validate up to 100 records in a BAM file:
$ pbvalidate --max-records 100 in.subreads.bam
To validate up to 100 records in a BAM file (equivalent to --max-
records=100
):
$ pbvalidate --quick in.subreads.bam
To validate a BAM file, using a specified log level:
$ pbvalidate --log-level=INFO in.subreads.bam
To validate a BAM file and write log messages to a file rather than to
stdout:
$ pbvalidate --log-file validation_results.log in.subreads.bam
sawriter
The sawriter tool generates a suffix array file from an input FASTA file. It
is used to prebuild suffix array files for reference sequences which can
later be used in resequencing workflows.
sawriter comes with blasr,
and is independent of
python.
Usage
sawriter saOut fastaIn [fastaIn2 fastaIn3 ...] [-blt p] [-larsson] [-4bit] [-manmy]
[-kar]
or
sawriter fastaIn (writes to fastIn.sa)
Options Description
-blt p Builds a lookup table on prefixes of length p. This speeds up lookups
considerably (more than the LCP table), but misses matches less than p
when searching.
-4bit Reads in one FASTA file as a compressed sequence file.
-larsson Uses the Larsson and Sadakane method to build the array. (Default)
-mamy Uses the MAnber and MYers method to build the array. This is slower than
the Larsson method, and produces the same result. This is mainly for
double-checking the correctness of the Larsson method.

Page 82
summarize
Modifications
The summarizeModifications tool generates a GFF summary file
(
alignment_summary.gff) from the output of base modification analysis
(i.e.
ipdSummary) combined with the coverage summary GFF generated
by resequencing pipelines. This is useful for power users running custom
workflows.
Usage
summarizeModifications [-h] [--version] [--emit-tool-contract]
[--resolved-tool-contract RESOLVED_TOOL_CONTRACT]
[--log-file LOG_FILE]
[--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL} | --debug
| --quiet | -v]
modifications alignmentSummary gff_out
Input Files
• modifications: Base Modification GFF file.
• alignmentSummary: Alignment Summary GFF file.
Output Files
• gff_out: Coverage summary for regions (bins) spanning the reference
with Base Modification results for each region
.
-kark Uses the Karkkainen DS3 method for building the suffix array. This is
probably slower than the Larsson method, but takes only
N/(sqrt 3)extra space.
-welter Uses lightweight suffix array construction. This is a bit slower than the
normal Larsson method.
-welterweight N Uses a difference cover of size N for building the suffix array. Valid values
are 7, 32, 64, 111, and 2281.
Options Description
Options Description
-h, --help Displays help information and exits.
--version Displays program version number and exits.
--emit-tool-contract Outputs the tool contract to stdout. (Default = False)
--resolved-tool-contract
RESOLVED_TOOL_CONTRACT
Runs the tool directly from a PacBio Resolved tool contract.
(Default = None)
--log-file LOG_FILE Writes the log to file. Default (None) will write to stdout.
--log-level Specifies the log level; values are [DEBUG, INFO, WARNING, ERROR,
CRITICAL] (Default = INFO)
--debug Alias for setting the log level to DEBUG. (Default = False)
--quiet Alias for setting the log level to CRITICAL to suppress output.
(Default = False)
--verbose, -v Sets the verbosity level. (Default = None)

Page 83
Appendix A - Application Entry Points and Output Files
Assembly
(HGAP 4)
Analysis Application Name: cromwell.workflows.pb_hgap4
Entry Point
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
Key Output Files
Base
Modification
Detection
Analysis Application Name: cromwell.workflows.pb_basemods
Entry Points
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
Key Output Files
Circular
Consensus
Sequencing
(CCS)
Analysis Application Name: cromwell.workflows.pb_ccs
Entry Point
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
Key Output Files
File Name Datastore SourceId
Coverage Summary pb_hgap4.coverage_gff
Alignments pb_hgap4.mapped
Polished Assembly pb_hgap4.consensus_fasta
Polished Assembly pb_hgap4.consensus_fastq
Draft Assembly pb_hgap4.ofile_a_ctg_fasta, pb_hgap4.ofile_p_ctg_fasta
File Name Datastore SourceId
Motifs and Modifications pb_basemods.motifs_gff
Motifs Summary pb_basemods.motifs_csv
Full Kinetics Summary pb_basemods.basemods_gff
IPD Ratios pb_basemods.basemods_csv
File Name Datastore SourceId
FASTQ file ccs_fastq_out
FASTA file ccs_fasta_out

Page 84
CCS with
Mapping
Analysis Application Name: cromwell.workflows.pb_ccs_mapping
Entry Points
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
Key Output Files
Convert BAM to
FASTX
Analysis Application Name: cromwell.workflows.pb_bam2fastx
Entry Point
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
Key Output Files
BAM file ccs_bam_out
Consensus Sequences pb_ccs.ccsxml
CCS Statistics pb_ccs.report_ccs
File Name Datastore SourceId
File Name Datastore SourceId
Coverage Summary pb_ccs_mapping.coverage_gff
Alignments pb_ccs_mapping.mapped
FASTQ file ccs_fastq_out
FASTA file ccs_fasta_out
BAM file ccs_bam_out
Consensus Sequences pb_ccs_mapping.ccsxml
CCS Statistics pb_ccs_mapping.report_ccs
Aligned BAM pb_ccs_mapping.mapped_bam
BAM Index pb_ccs_mapping.mapped_bam_bai
File Name Datastore SourceId
FASTQ file(s) pb_bam2fastx.fastq_zip
FASTA file(s) pb_bam2fastx.fasta_zip

Page 85
Demultiplex
Barcodes
Analysis Application Name: cromwell.workflows.pb_demux_subreads
Entry Points
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
:id: eid_barcode
:name: Entry eid_barcode
:fileTypeId: PacBio.DataSet.BarcodeSet
Key Output Files
Demultiplex
Barcodes
(CCS-Only)
Analysis Application Name: cromwell.workflows.pb_demux_ccs
Entry Points
:id: eid_ccs
:name: Entry eid_ccs
:fileTypeId: PacBio.DataSet.ConsensusReadSet
:id: eid_barcode
:name: Entry eid_barcode
:fileTypeId: PacBio.DataSet.BarcodeSet
Key Output Files
Iso-Seq
Analysis
Analysis Application Name: cromwell.workflows.pb_isoseq3
Entry Points
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
:id: eid_barcode
:name: Entry eid_barcode
:fileTypeId: PacBio.DataSet.BarcodeSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
File Name Datastore SourceId
Barcode Report Details pb_demux_subreads.summary_csv
Demultiplexed Datasets Pb_demux_subreads.barcoded_reads
Unbarcoded Reads Pb_demux_subreads.unbarcoded
File Name Datastore SourceId
Barcode Report Details pb_demux_ccs.summary_csv
Demultiplexed Datasets Pb_demux_ccs.barcoded_reads
Unbarcoded Reads Pb_demux_ccs.unbarcoded
Page 86
Key Output Files
Iso-Seq
Analysis
(CCS-Only)
Analysis Application Name: cromwell.workflows.pb_isoseq3_ccsonly
Entry Points
:id: eid_ccs
:name: Entry eid_ccs
:fileTypeId: PacBio.DataSet.ConsensusReadSet
:id: eid_barcode
:name: Entry eid_barcode
:fileTypeId: PacBio.DataSet.BarcodeSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
Key Output Files
File Name Datastore SourceId
Collapsed Filtered Isoforms FASTQ pb_isoseq3.collapse_fastq
Collapsed Filtered Isoforms GFF pb_isoseq3.collapse_gff
Group TXT pb_isoseq3.collapse_group
Abundance TXT pb_isoseq3.collapse_abundance
Read Stat TXT pb_isoseq3.collapse_readstat
High-Quality Transcripts pb_isoseq3.hq_fastq
Low-Quality Transcripts pb_isoseq3.lq_fastq
CCS FASTQ pb_isoseq3.ccs_fastq_zip
Full-length CCS pb_isoseq3.flnc_bam
Polished Report pb_isoseq3.polish_report_csv
Cluster Report pb_isoseq3.report_isoseq
File Name Datastore SourceId
Collapsed Filtered Isoforms FASTQ pb_isoseq3_ccsonly.collapse_fastq
Collapsed Filtered Isoforms GFF pb_isoseq3_ccsonly.collapse_gff
Group TXT pb_isoseq3_ccsonly.collapse_group
Abundance TXT pb_isoseq3_ccsonly.collapse_abundance
Read Stat TXT pb_isoseq3_ccsonly.collapse_readstat
High-Quality Transcripts pb_isoseq3_ccsonly.hq_fastq
Low-Quality Transcripts pb_isoseq3_ccsonly.lq_fastq
CCS FASTQ pb_isoseq3_ccsonly.ccs_fastq_zip
Full-length CCS pb_isoseq3._ccsonly.flnc_bam
Polished Report pb_isoseq3._ccsonly.polish_report_csv
Cluster Report pb_isoseq3._ccsonly.report_isoseq

Page 87
Long Amplicon
Analysis (LAA)
Analysis Application Name: cromwell.workflows.pb_laa
Entry Point
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
Key Output Files
Mapping
Analysis Application Name: cromwell.workflows.pb_align_ccs
Entry Points
:id: eid_ccs
:name: Entry eid_ccs
:fileTypeId: PacBio.DataSet.ConsensusReadSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
Key Output Files
Microbial
Assembly
Analysis Application Name:
cromwell.workflows.pb_assembly_microbial
Entry Point
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
Key Output Files
File Name Datastore SourceId
Consensus Sequence Statistics CSV pb_laa.summary_csv
Chimeric/Noise Consensus
Sequences
pb_laa.chimeras_fastq
Consensus Sequences pb_laa.consensus_fastq
Consensus Sequences by Barcode pb_laa.consensus_fastq_split
Chimeric/Noise Consensus
Sequences by Barcode
pb_laa.chimeras_fastq_split
File Name Datastore SourceId
Mapped reads pb_align_ccs.mapped
Coverage summary pb_align_ccs.coverage_gff
File Name Datastore SourceId
Polished assembly pb_assembly_microbial.consensus_fasta/fastq
Polished Contigs After oriC Rotation pb_assembly_microbial.assembled_fasta
Coverage Summary pb_assembly_microbial.coverage_gff
Final Assembly pb_assembly_microbial.ncbi_fasta

Page 88
Minor Variants
Analysis
Analysis Application Name: cromwell.workflows.pb_mv_ccs
Entry Points
:id: eid_ccs
:name: Entry eid_ccs
:fileTypeId: PacBio.DataSet.ConsensusReadSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
Key Output Files
Resequencing
Analysis Application Name: cromwell.workflows.pb_resequencing
Entry Points
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
Key Output Files
Mapped BAM pb_assembly_microbial.mapped
File Name Datastore SourceId
File Name Datastore SourceId
Minor Variants HTML Reports pb_mv_ccs.juliet_html
Per-Variant Table pb_mv_ccs.report_csv
Alignments pb_mv_ccs.mapped
File Name Datastore SourceId
Coverage and Variant Call Summary pb_resequencing.consensus_gff
Variant Calls pb_resequencing.variants_gff
Consensus Contigs pb_resequencing.consensus_fastq
Variant Calls pb_resequencing.variants_vcf
Alignments pb_resequencing.mapped
Coverage Summary pb_resequencing.coverage_gff
Consensus Sequences pb_resequencing.consensus_fasta
Aligned BAM pb_resequencing.mapped_bam
BAM Index pb_resequencing.mapped_bam_bai

Page 89
Site
Acceptance
Test (SAT)
Analysis Application Name: cromwell.workflows.pb_sat
Entry Points
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
Key Output Files
Structural
Variant Calling
Analysis Application Name: cromwell.workflows.pb_sv_clr
Entry Points
:id: eid_subread
:name: Entry eid_subread
:fileTypeId: PacBio.DataSet.SubreadSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
Key Output Files
Structural
Variant Calling
(CCS-Only)
Analysis Application Name: cromwell.workflows.pb_sv_ccs
Entry Points
:id: eid_ccs
:name: Entry eid_ccs
:fileTypeId: PacBio.DataSet.ConsensusReadSet
:id: eid_ref_dataset
:name: Entry eid_ref_dataset
:fileTypeId: PacBio.DataSet.ReferenceSet
File Name Datastore SourceId
Coverage and Variant Call Summary pb_sat.consensus_gff
Variant Calls pb_sat.variants_gff
Consensus Contigs pb_sat.consensus_fastq
Variant Calls pb_sat.variants_vcf
Alignments pb_sat.mapped
Coverage Summary pb_sat.coverage_gff
Consensus Sequences pb_sat.consensus_fasta
File Name Datastore SourceId
Structural Variants pb_sv_clr.variants
Aligned reads
(BioSampleName)
pb_sv_clr.alignments_by_sample_datastore

Page 90
Key Output Files
File Name Datastore SourceId
Structural Variants pb_sv_ccs.variants
Aligned reads
(BioSampleName)
pb_sv_ccs.alignments_by_sample_datastore

Page 91
Appendix B - Third Party Command-Line Tools
Following is information on the third-party command-line tools included in
the
smrtcmds/bin subdirectory.
bamtools
• A C++ API and toolkit for reading, writing, and manipulating BAM files.
• See https://sourceforge.net/projects/bamtools/ for details.
cromwell
• Scientific workflow engine used to power SMRT Link.
• See https://cromwell.readthedocs.io/en/stable/ for details.
daligner,
LAsort,
LAmerge,
HPC.daligner
• Finds all significant local alignments between reads.
• See https://dazzlerblog.wordpress.com/command-guides/daligner-
command-reference-guide/ for details.
datander
• Finds all local self-alignment between long, noisy DNA reads.
• See https://github.com/thegenemyers/DAMASKER for details.
DB2fasta,
DBdump,
DBdust, DBrm,
DBshow,
DBsplit,
DBstats,
Fasta2DB
Utilities that work with Dazzler databases:
• DB2fasta: Converts database files to FASTA format.
• DBdust: Runs the DUST algorithm over the reads in the untrimmed
database, producing a track that marks all intervals of low complexity
sequence.
• DBdump/DBshow: Displays a subset of the reads in the database;
selects the information to show about the reads, including any mask
tracks.
• DBrm: Deletes all the files in a given database.
• DBsplit: Divides a database conceptually into a series of blocks.
• DBstats: Shows overview statistics for all the reads in the trimmed
database.
• Fasta2DB: Builds an initial database, or adds to an existing database,
using a list of
.fasta files.
• See https://dazzlerblog.wordpress.com/command-guides/dazz_db-
command-guide/ for details.
ipython
• An interactive shell for using the Pacific Biosciences API.
• See https://ipython.org/ for details.
python
• An object-oriented programming language.
• See https://www.python.org/ for details.
REPmask,
TANmask,
HPC.REPmask,
HPC.TANmask
• A set of programs to soft-mask all tandem and interspersed repeats in
Dazzler databases when computing overlaps.
• See https://github.com/thegenemyers/DAMASKER for details.

Page 92
samtools
• A set of programs for interacting with high-throughput sequencing data
in SAM/BAM/VCF formats.
• See http://www.htslib.org/ for details.

Page 93
Appendix C - Microbial Assembly Advanced Options
Use this application to generate de novo assemblies of small prokaryotic
genomes between 1.9-10 Mb and companion plasmids between 2 – 220
kb.
The Microbial Assembly application:
• Includes chromosomal- and plasmid-level de novo genome assembly,
circularization, polishing, and rotation of the origin of replication for
each circular contig.
• Facilitates assembly of larger genomes (yeast) as well.
• Accepts Sequel data (BAM format) as input.

Page 94
The workflow shown above consists of two assembly stages:
Stage 1: Intended for contig assembly of large sequences. This stage uses
the seed length cutoff which might miss small sequences in the input
sample (smaller than the input cutoff, such as the plasmids).
Stage 2: Intended for a fine-grained assembly. This stage assembles only
the unmapped and poorly mapped reads, does not use a seed length
cutoff, and relaxes the overlapping parameters.
Available options for these two stages are identical. The only differences
are:
1. Stage 1 parameters are prefixed with stage1 and Stage 2 parameters
with
stage2.
2. Default values.
Complete list of all available options and their default values
genome_size = 5000000
coverage = 30
plasmid_contig_len_max = 300000
plasmid_min_aln_frac = 0.95
plasmid_dedup_min_frac = 0.90
stage1.length_cutoff = -1
stage1.block_size = 1024
stage1.use_median_filter = 1
stage1.ovl_opt_raw =
stage1.ovl_opt_erc =
stage1.ovl_flank_grace = 20
stage1.ovl_min_idt = 96
stage1.ovl_min_len = 1000
stage1.ovl_filter_opt = --max-diff 80 --max-cov 100 --min-cov 1 --bestn 20 --min-len
4000 --gapFilt --minDepth 4
stage2.length_cutoff = 0
stage2.block_size = 1024
stage2.use_median_filter = 1
stage2.ovl_opt_raw = --min-map-len 499
stage2.ovl_opt_erc = --min-map-len 499
stage2.ovl_flank_grace = 20
stage2.ovl_min_idt = 94
stage2.ovl_min_len = 500
stage2.ovl_filter_opt = --max-diff 10000 --max-cov 10000 --min-cov 1 --bestn 20 --min-
len 498 --gapFilt --minDepth 4

Page 95
Advanced Parameters Default Value Description
stage1.length_cutoff -1 Only reads as long as this value will be used as seeds in the
draft assembly, and subsequently error-corrected.
-1 means this will be calculated automatically so that the total
number of seed bases equals (Genome Length x Coverage).
0 means all reads in the input Data Set will be used for error-
correction.
stage1.block_size 1024 The overlapping process is performed on pairs of blocks of
input sequences, where each block contains the number of
sequences which crop up to this size (in Mbp). Note: The
number of pairwise comparisons grows quadratically with the
number of blocks (meaning more cluster jobs), but also the
larger the block size the more resources are required to
execute each pairwise comparison.
stage1.use_median_filter 1 The median filter selects one subread per ZMW – the median
length subread. 1 enables the filter, while 0 deactivates it. It is
highly recommended to use the median filter.
stage1.ovl_opt_raw NONE Overlapping options for the Raptor overlapping tool, applied
at the raw read overlapping stage (pre-assembly). The
defaults are set to work well with PacBio subreads. The options
set by this parameter here are fed directly into the Raptor call.
For details on Raptor options, use raptor -h.
stage1.ovl_opt_erc NONE Overlapping options for the Raptor overlapping tool, applied
at the pread overlapping stage. The defaults are set to work
well with error-corrected reads and HiFi reads. The options set
by this parameter here are fed directly into the Raptor call.
For details on Raptor options, use raptor -h.
stage1.ovl_flank_grace 20 Heuristic to salvage some potential dovetail overlaps. Only
dovetail overlaps are used for assembly, and all other overlaps
(partial overlaps, which are actually local alignments by
definition) are not used to construct the string graph.
Dovetail overlaps are overlaps where the full suffix of one read
and a full prefix of the other read are used to form the overlap.
More details can be found here:
http://wgs-assembler.sourceforge.net/wiki/index.php/Overlaps
Overlaps are formed in the process of alignments, and
alignment extension near the ends of the sequences can be
stopped in case there are errors present near the edges of one
or both of the sequences.
For any overlap which is missing only a few bases to become a
dovetail overlap (the number of bases defined by this
parameter), the coordinates are augmented to convert it into a
dovetail overlap.
The impact of this parameter is very low, and this value is set to
work in almost all cases. This value should also be set
relatively low, to avoid chimeric overlaps.
stage1.ovl_min_idt 96 Overlap identity threshold (in percentage) for filtering overlaps
used for contig construction.
stage1.ovl_min_len 1000 Minimum span of an overlap to keep it for contig construction,
in bp.

Page 96
stage1.ovl_filter_opt --max-diff
80 --max-cov
100 --min-
cov 1 --
bestn 20 --
min-len 4000
--gapFilt --
minDepth 4
Overlap filter options. These are identical to FALCON overlap
filtering options except for the addition of the two options listed
in the defaults:
--gapFilt - Enables the chimera filter, which analyzes each
pread's overlap pile, and determines whether a pread is
chimeric based on the local coverage across the pread.
--minDepth - Option for the chimera filter. The chimera filter
is ignored when a local region of a read has coverage lower
than this value.
The other parameters are:
--min-cov - Minimum allowed coverage at either the 5' or
the 3' end of a read. If the coverage is below this value, the
read is blacklisted and all of the overlaps it is incident with are
ignored. This helps remove potentially chimeric reads.
--max-cov - Maximum allowed coverage at either the 5' or
the 3' end of a read. If the coverage is above this value, the
read is blacklisted and all of the overlaps it is incident with are
ignored. This helps remove repetitive reads which can make
hairballs in the string graph. Note that this value is a heuristic
which works well for ~30x seed length cutoff. If the cutoff is set
higher, it is advised that this value is also increased.
--max-diff - Maximum allowed difference between the
coverages at the 5' and 3' ends of any particular read. If the
coverage is above this value, the read is blacklisted and all of
the overlaps it is incident with are ignored.
--bestn - Keep at most this many overlaps on the 5' and the
3' side of any particular read.
--min-len - Filter overlaps where either A-read or the B-read
are shorter than this value.
stage2.length_cutoff 0 Only reads as long as this value will be used as seeds in the
draft assembly, and subsequently error-corrected. -1 means
this will be calculated automatically so that the total number of
seed bases equals (Genome Length x Coverage).
0 means all reads in the input Data Set will be used for error-
correction.
stage2.block_size 1024 The overlapping process is performed on pairs of blocks of
input sequences, where each block contains the amount of
sequences which crop up to this size (in Mbp). Note: The
number of pairwise comparisons grows quadratically with the
number of blocks (meaning: more cluster jobs), but also the
larger the block size the more resources are required to
execute each pairwise comparison.
stage2.use_median_filter 1 The median filter selects one subread per ZMW – the median
length subread. 1 enables the filter, while 0 deactivates it. It is
highly recommended to use the median filter.
stage2.ovl_opt_raw --min-map-
len 499
Overlapping options for the Raptor overlapping tool, applied
at the raw read overlapping stage (pre-assembly). The
defaults are set to work well with PacBio subreads. The options
set by this parameter here are fed directly into the Raptor call.
For details on Raptor options, use raptor -h.
The option --min-map-len reduces the minimum span of the
overlap to 499 bp (instead of the default 1000 bp). This allows
shorter overlaps to be reported.
Advanced Parameters Default Value Description

Page 97
stage2.ovl_opt_erc --min-map-
len 499
Overlapping options for the Raptor overlapping tool, applied
at the pread overlapping stage. The defaults are set to work
well with error-corrected reads and HiFi reads. The options set
by this parameter here are fed directly into the Raptor call.
For details on Raptor options, use raptor -h.
The option --min-map-len reduces the minimum span of the
overlap to 499 bp (instead of the default 1000 bp). This allows
shorter overlaps to be reported.
stage2.ovl_flank_grace 20 Heuristic to salvage some potential dovetail overlaps. Only
dovetail overlaps are used for assembly, and all other overlaps
(partial overlaps, which are actually local alignments by
definition) are not used to construct the string graph.
Dovetail overlaps are overlaps where the full suffix of one read
and a full prefix of the other read are used to form the overlap.
More details can be found here:
http://wgs-assembler.sourceforge.net/wiki/index.php/Overlaps
Overlaps are formed in the process of alignments, and
alignment extension near the ends of the sequences can be
stopped in case there are errors present near the edges of one
or both of the sequences.
For any overlap which is missing only a few bases to become a
dovetail overlap (the number of bases defined by this
parameter), the coordinates are augmented to convert it into a
dovetail overlap.
The impact of this parameter is very low, and this value is set to
work in almost all cases. This value should also be set
relatively low, to avoid chimeric overlaps.
stage2.ovl_min_idt 94 Overlap identity threshold (in percentage) for filtering overlaps
used for contig construction.
stage2.ovl_min_len 500 Minimum span of an overlap to keep it for contig construction,
in bp.
genome_size 5,000,000 The approximate number of base pairs expected in the
genome, used to determine the coverage cutoff.
Note: It is better to slightly overestimate rather than
underestimate the genome length to ensure good coverage
across the genome.
Coverage 30 A target value for the total amount of subread coverage used
for assembly. This parameter is used, together with the
genome size, to calculate the seed length cutoff.
plasmid_contig_len_max 300,000 Maximum expected plasmid size in the input subreadset. The
default value covers a large range of possible plasmids. This
value is used to select subreads for the secondary assembly
stage which is specialized for assembly of smaller sequences
(e.g. plasmids) that might have been lost due to the seed
length cutoff threshold.
Any contig assembled in the first assembly stage larger than
this value will be filtered out and reassembled in the secondary
assembly stage. This is performed in order to avoid partially
assembled plasmid sequences
plasmid_min_aln_frac 0.95 Applied in the "Mapping and filtering" stage, where raw
subreads are aligned to the filtered contigs of the first assembly
stage.
Any subread which doesn't have at least this large of aligned
span (in query coordinates) is kept for the secondary assembly
stage, in addition to all reads which didn't align)
The value is a fraction of the subread's length (0.95 means
95% of the subread's size).
Advanced Parameters Default Value Description

Page 98
For Research Use Only. Not for use in diagnostic procedures. © Copyright 2017 - 2019, Pacific Biosciences of
California, Inc. All rights reserved. Information in this document is subject to change without notice. Pacific Biosciences
assumes no responsibility for any errors or omissions in this document. Certain notices, terms, conditions and/or use
restrictions may pertain to your use of Pacific Biosciences products and/or third party products. Please refer to the
applicable Pacific Biosciences Terms and Conditions of Sale and to the applicable license terms at
https://www.pacb.com/legal-and-trademarks/terms-and-conditions-of-sale/.
Pacific Biosciences, the Pacific Biosciences logo, PacBio, SMRT, SMRTbell, Iso-Seq and Sequel are trademarks of
Pacific Biosciences. BluePippin and SageELF are trademarks of Sage Science, Inc. NGS-go and NGSengine are
trademarks of GenDx. FEMTO Pulse and Fragment Analyzer are trademarks of Agilent Technologies Inc. All other
trademarks are the sole property of their respective owners.
See https://github.com/broadinstitute/cromwell/blob/develop/LICENSE.txt for Cromwell redistribution information.
P/N 100-939-900 Version 09 (November 2019)
plasmid_dedup_min_frac 0.90 Applied in the "Deduplicate plasmid contigs" stage, where
contigs from the secondary assembly stage are aligned to the
contigs of the first assembly stage. This is done because
reusing unmapped and poorly mapped reads can still cause
duplicate contigs to form in the secondary assembly stage.
After contigs from the secondary stage are aligned, any contig
whose alignment doesn't cover at least this fraction of it's
length is kept. All other contigs are marked as duplicates and
removed.
Advanced Parameters Default Value Description
