1
Transposable elements drive the evolution of metazoan zinc finger genes
Jonathan N. Wells
1
, Ni-Chen Chang
1,*
, John McCormick
1,*
, Caitlyn Coleman
2
, Nathalie Ramos
1,3
,
Bozhou Jin
1
, Cédric Feschotte
1
1. Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14850, USA.
2. Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida,
Tampa, FL 33620, USA.
3. Department of Genetics and Genomic Sciences, Center for Transformative Disease Modeling,
Tisch Cancer Institute, Icahn Institute for Data Science and Genomic Technology, Icahn School
of Medicine at Mount Sinai, New York, NY 10029, USA.
* These authors contributed equally to this work.
Abstract
Cys2-His2 Zinc finger genes (ZNFs) form the largest family of transcription factors in metazoans. ZNF
evolution is highly dynamic and characterized by the rapid expansion and contraction of numerous
subfamilies across the animal phylogeny. The forces and mechanisms underlying rapid ZNF evolution
remain poorly understood, but there is growing evidence that the targeting and repression of lineage-
specific transposable elements (TEs) plays a major role in the diversification of the Kruppel-associated
box ZNF (KZNF) subfamily, which predominates in tetrapod genomes. At present, it is unknown whether
this function and co-evolutionary relationship is unique to KZNFs, or a broader feature of metazoan
ZNFs. Here, we present evidence that genomic conflict with TEs has been a central driver in the
diversification of ZNFs in animals. Sampling from more than 4000 animal genome assemblies, we show
that the copy number of retroelements correlates with that of ZNFs across at least 750 million years of
metazoan evolution, both within and between major taxonomic groups. Using computational predictions,
we show that ZNFs preferentially bind TEs in a diverse set of representative animal species. We further
investigate one of the most expansive ZNF subfamilies found in cyprinid fish, which are characterized by
a conserved domain we dubbed the Fish N-terminal Zinc-finger associated (FiNZ) domain. FiNZ-ZNFs
have dramatically expanded in several fish species, including the zebrafish in which we predict ~700
FiNZ-ZNF genes. Almost all are located on the long arm of chromosome 4, and recent duplicates are
evolving adaptively under positive selection. Like mammalian KZNFs, the bulk of zebrafish FiNZ-ZNFs
are expressed in waves at the onset of zygotic genome activation. Blocking FiNZ-ZNF translation using
morpholinos during early zebrafish embryogenesis results in a global de-repression of young,
transcriptionally active TEs, likely driven by the failure to establish heterochromatin over these elements.
Together, these data suggest that ZNF diversification has been intimately connected to TE expansion
throughout animal evolution and that families of ZNFs have been deployed independently in fish and
mammals to repress TEs during early embryogenesis.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
2
Introduction
The Cys2-His2 Zinc Finger (ZNF) is a short nucleic acid binding domain widespread in eukaryotes. ZNFs
are often arranged in tandemly repeated arrays, resulting in proteins with versatile nucleic acid binding
ability (Wolfe et al., 2000; Najafabadi et al., 2017; Klug, 2010). In metazoans tandem-repeat ZNF genes
have undergone dramatic lineage-specific expansions in gene copy number, as well as an increase in the
diversity of DNA sequences that they recognize (Heger et al., 2020; Najafabadi et al., 2017). Many ZNF
proteins contain accessory domains that modulate chromatin states and transcription activity, such as the
repressive Krüppel-associated box (KRAB) (Bellefroid et al., 1991). Thus, ZNFs represent the largest
and most dynamically evolving transcription factor family in metazoans.
The abundance and diversity of C2H2 ZNF genes is a hallmark feature of metazoan genomes (Najafabadi
et al., 2017; Heger et al., 2020), and in species whose genomes have been sequenced, ZNF gene copy
number spans three orders of magnitude, from fewer than ten in many nematodes, to hundreds, if not
thousands in vertebrates, arthropods, and mollusks (Albertin et al., 2015; Goodstadt et al., 2007; Panfilio
et al., 2019). Their lineage-specificity can be seen through comparisons of closely related species. For
example, a survey of three great ape species and rhesus macaque found that approximately 10% (213 out
of 2,253) of predicted KRAB-ZNFs (KZNFs) were species-specific, including 7 from humans and 145 in
orangutangs (Nowick et al., 2011). This rapid evolution contrasts with most other transcription factors,
which are deeply conserved across metazoans (Degnan et al., 2009).
However, the same qualities that differentiate ZNFs from other transcription factors make them
challenging to study, and consequently there have been few attempts to produce a unified theory
explaining their prevalence and recurrent expansions (Emerson and Thomas, 2009; Fedotova et al., 2017).
Early hints at the function of the KZNFs came from work showing that deletion of the genes TRIM28 and
the K3K9 methyltransferase SETDB1 led to dramatic increases in endogenous retrovirus expression in
mice (Rowe et al., 2010; Matsui et al., 2010). Since TRIM28 was known to be an important cofactor of
the KRAB domain, this led to the proposal that KZNFs are in a coevolutionary relationship with
retroelements (Thomas and Schneider, 2011). In this study, we aim to test whether genetic conflict with
transposable elements (TEs) is the central force driving the rapid evolution of metazoan ZNFs more
generally.
TEs are selfish genetic elements that replicate independently within their host genomes, and in most
eukaryotes comprise between 5% and 85% of the genome. While individual TE insertions are most likely
to be neutral or deleterious, in aggregate, TEs represent an important source of genetic variation and
innovation. For example, in vertebrates TEs are a major source of cis-regulatory elements and coding
sequences coopted for host gene regulation (Senft and Macfarlan, 2021; Fueyo et al., 2022; Almeida et
al., 2022). On the other hand, uncontrolled TE proliferation has deleterious consequences ranging from
insertional mutagenesis and genomic instability caused by ectopic recombination between TE insertions,
to dysregulation of gene expression stemming from the fact that many TEs carry their own promoters and
regulatory sequences. Consequently, metazoans have evolved a variety of defenses to control their spread,
most notably the piRNA system (Aravin et al., 2007; Czech and Hannon, 2016) and the previously
mentioned KZNFs (Ecco et al., 2017; Bruno et al., 2019). One hallmark of both of these defense systems
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
3
is their ability to adapt to changing population of invaders, which leads to selection pressure on the
invaders to evade these defenses, thereby creating arms race dynamics (McLaughlin and Malik, 2017).
There is growing evidence that KZNFs are engaged in such arms races with TEs (Bruno et al., 2019;
Fernandes et al., 2018; Jacobs et al., 2014). KZNFs form the largest ZNF gene subfamily found in
Sarcopterygii, i.e. tetrapods and lobe-finned fish (Imbeault et al., 2017; Bellefroid et al., 1991). The
extensive variation in KZNF copy number across species has long been appreciated (Bellefroid et al.,
1991; Huntley et al., 2006), but the evolutionary forces driving this remained elusive until a breakthrough
study showing that the number of ZNF domains in a given genome is positively correlated to retroelement
copy number across a small but diverse sample of vertebrates, suggesting a coevolutionary relationship
between the two (Thomas and Schneider, 2011). This relationship was later bolstered by ChIP-seq
experiments in humans and mice mapping the genome-wide binding of hundreds of KZNFs, which
revealed that most target specific TE families (Imbeault et al., 2017; Wolf et al., 2020). Furthermore,
KZNF knockouts in mouse and humans lead to upregulation of TE expression (Haring et al., 2021; Wolf
et al., 2020). Mechanistic studies showed that TE transcriptional repression via KZNF proteins is
typically mediated via their KRAB domain, which interacts with KAP1/TRIM28 corepressor to recruit
the H3K9me3 writer, SETDB1, amongst several other chromatin silencing factors (Wolf and Goff, 2009;
Rowe et al., 2010; Matsui et al., 2010; Ecco et al., 2017); in doing so, most KZNFs nucleate the formation
of heterochromatin at their target TE loci. Together, these findings support the idea that TE proliferation
is a driving force behind the diversification of KZNFs.
Much less is known about the factors driving the evolution of other ZNF families. KZNFs represent a
small fraction of all ZNFs, being restricted to some thirty thousand tetrapods, against a background of
millions of animal species (Mora et al., 2011; Sahney et al., 2010), most of which are predicted to harbor
hundreds of individual ZNF genes. One of the few other large families to have been studied are the zinc
finger-associated domain (ZAD)-ZNFs, which are found in many insect species (Chung et al., 2007). The
handful of ZAD-ZNFs characterized in Drosophila melanogaster do not appear to have clear roles in TE
repression, but instead perform a variety of functions related to heterochromatin organization (Kasinathan
et al., 2020). Thus far, only one ZAD-ZNF gene (CG17801) has been reported to directly affect TE
transcript levels, and was identified as part of an earlier screen for piRNA pathway components (Czech et
al., 2013). More recently, the ZAD-ZNF protein Kipferl has been shown to regulate TE activity indirectly,
by targeting the protein Rhino to heterochromatic piRNA clusters in Drosophila melanogaster, thus
licensing piRNA production from these loci (Baumgartner et al., 2022). Despite apparent differences in
the immediate function of most ZAD- and KZNF genes, both families share features that are broadly
characteristic of the majority of ZNF genes thus far studied, namely: rapid turnover and sequence
evolution, evidence of being under positive selection, involvement in establishing or maintaining
heterochromatin, and expression during early embryogenesis (Chung et al., 2007; Kasinathan et al., 2020;
Ecco et al., 2017). Other highly expanded ZNF families, such as those of hemipteran bugs, octopus, and
zebrafish, also possess subsets of these features (Panfilio et al., 2019; Albertin et al., 2015; Howe et al.,
2016).
Here, we investigate the hypothesis that interaction with TEs is a driving force underlying the molecular
evolution and functional diversification of ZNF families across metazoans. Through a survey of all
currently available metazoan genome assemblies, we find that the number of ZNF open reading frames is
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
4
positively correlated with the number of retroelements across a large and diverse sample of animals.
Using in silico predictions of ZNF binding specificity, we show that ZNFs from a sample of distantly
related animals preferentially recognize TEs, as has been shown for KZNFs. We then turn our focus to a
large family of ZNFs present in zebrafish, which are defined by its association with a conserved protein
domain we have termed Fish N-terminal Zinc-finger associated (FiNZ). These genes are unique to
cyprinid fish and have evolved in parallel to the KZNFs found in tetrapods. Using refined gene
annotations, we find that in zebrafish, recently duplicated FZNF paralogs are under positive selection,
consistent with ongoing arms races with TEs. Finally, by simultaneously targeting over 400 FZNF genes
for knockdown during zebrafish embryogenesis, we show that FZNF depletion causes de-repression of
TEs.
Results
Annotation of ZNFs and TEs in metazoan genomes
We sought to estimate the number of ZNF genes in a large set of publicly available metazoan genome
assemblies. ZNF genes are challenging to annotate due to their highly repetitive nature and restricted
patterns of mRNA expression. Consequently, even in well curated genome assemblies, the true number of
functional ZNF genes is likely underestimated (Huntley et al., 2006; White et al., 2017). To avoid biases
caused by incomplete gene annotation, we used profile-HMM searches with HMMER (Eddy, 2011) to
identify ZNF open reading frames (ORFs), here defined as any stretch of protein coding sequence,
regardless of whether they are initiated by a start codon. As our aim in this project was to investigate
tandem-repeat ZNF genes, we restricted our search to ORFs with at least five ZNF domains. Searching
3,221 metazoan genome assemblies revealed extensive variation across species, from fewer than ten ZNF
ORFs in most nematode worms, to upwards of a thousand in many vertebrates, mollusks and arthropods
(Fig 1A, Supp. Data 1-2). Since estimates of ZNFs could be biased by genome assembly quality, we set a
minimum scaffold N50 of 50 kb to filter out low quality assemblies. To ensure that this threshold was
appropriate we calculated the correlation between our ZNF ORFs counts and scaffold N50 length (Supp.
Fig. 1A). This revealed a significant, but weak correlation of rho = 0.1, reassuring us that assembly
quality was not unduly biasing our ORF counts.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint

5
Figure 1. Annotation of ZNF ORFs and full genes in metazoan genome assemblies.
A) Boxplots showing distributions of ZNF ORF counts per species in each major taxonomic phylum.
Adjacent to boxplots are the number of representative species per phylum; *Number by nematodes
excludes those with zero tandem repeat ZNF ORFs (defined as containing at least 5 repeats). Phylogeny
acquired from TimeTree5 (Kumar et al., 2022). B) Maximum likelihood phylogenetic tree of
Cypriniformes species with high quality genome assemblies. Numbers represent counts of FZNF genes
using improved gene annotations. Nodes marked in blue have lower than 95/95 support for Shimodaira-
Hasegawa approximate likelihood ratio test/ultra-fast bootstraps, respectively.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
6
In addition to automated counts, we carried out more thorough gene annotations in cyprinid fish (Supp.
Data 2). Cyprinids, which include the widely used model organism Danio rerio along with many
economically and culturally important carp species, are one of the most species-rich vertebrate families,
with over 1,500 extant members (Fricke et al., 2020). They possess a large family of ZNF genes
characterized by the presence of a 28-amino acid N-terminal domain we dubbed the FiNZ domain (Supp.
Fig. 2A). The domain has no significant similarity to known protein domains but is highly conserved
across cyprinid ZNFs and enriched in negatively charged glutamic acid residues (p = 3e-06, relative to
sequences in the SwissProt51 database (Vacic et al., 2007)). The canonical structure of FinZ-ZNF
(FZNF) genes resembles that of KZNFs, with the FiNZ domain being encoded by a single exon upstream
of a second exon consisting of an array of ZNF domains (Imbeault et al., 2017). However, the number of
copies of the FiNZ domain contained in the first coding exon varies between species – for example, in
zebrafish the FiNZ exon almost always contains a single, stand-alone copy, whereas in goldfish it
contains up to four tandemly repeated copies.
Using an iterative approach, we annotated putative FZNF genes using a combination of blast (Altschul et
al., 1990), HMMER (Eddy, 2011) and Augustus-PPX (Hoff and Stanke, 2018; Keller et al., 2011). In
zebrafish, this approach yielded a total of 684 FZNF genes, a substantial increase from the number
annotated in the preexisting RefSeq and Ensembl gene sets (Supp. Fig. 2B). Remarkably, almost all
FZNF genes are located on the long arm of chromosome four, a region previously noted for its
enrichment in TEs, ZNF and immunity genes, as well as a wide variety of small RNA-encoding loci
(Howe et al., 2016; Howe et al., 2013). Across the cyprinid family, the copy number of predicted FZNF
genes varies substantially, even within closely related species (Fig. 1B); within the Danio genus alone,
the number of predicted FZNF genes varies by an order of magnitude. To further validate these copy
number estimates, we used raw reads from four Danio species and compared the read coverage over
FZNF genes to coverage over BUSCO (Benchmarking Universal Single-Copy Orthologs) genes. In three
out of four cases, there was good agreement between the two, but in the case of Danio albolineatus, the
median read depth over FZNF genes was approximately 50% greater than that of BUSCO genes (Supp.
Fig. 3A). This indicates that in many species, the true number of ZNF genes is likely to be far higher than
gene annotation pipelines can predict, highlighting the difficulty of assembling and accurately counting
these highly repetitive genes.
Finally, to verify that our automated ZNF ORF counts are good proxies for the number of annotated ZNF
genes in a species, we compared these counts to those of our reannotated FZNFs, as well as independent
estimates of KZNF copy number in tetrapods (Imbeault et al., 2017). In both cases, we found good
agreement between the simpler ORF counts and higher quality estimates based on gene annotations
(respectively, Spearman’s rho = 0.91, p-value = 2.6e-59; rho = 0.97, p-value = 1.7e-05; Supp. Fig. 3B, C).
This relationship is approximately linear, but ORF counts grow faster than annotated gene counts due to
factors such as genes having multiple exons and the presence of pseudogenes or gene fragments. Satisfied
that our gene annotations were of sufficient quality for further downstream analysis, we began to test for
evidence of coevolution between ZNFs and TEs.
Correlation between ZNFs and retroelements
Early evidence for a coevolutionary relationship between TEs and ZNFs was reported by Thomas and
Schneider, who observed a strong correlation between ZNF and retroelement copy number in a sample of
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
7
26 vertebrate species (Thomas and Schneider, 2011). By leveraging the greatly increased number of
currently available genome assemblies, we sought to reproduce and expand on these results. Using the
same process as for ZNF ORFs, we searched for reverse transcriptase and RNaseH domains (Supp. Data
1-2). These domains are highly conserved, and characteristic of proteins encoded by long terminal repeat
(LTR) and non-LTR retroelements, making their counts rough proxies for the number of autonomous
retroelement insertions in each genome.
A challenge with this approach is to address the issue of autocorrelation produced by the non-
independence of phylogenetically related species (Felsenstein, 1985). Since there is currently no well-
established phylogenetic tree that includes all sequenced metazoan genomes, we opted to select a single
species per taxonomic family, leveraging the fact that both ZNF and TE turnover is typically very rapid,
so that even closely related species often have largely independent sets of both (Liu et al., 2014; Imbeault
et al., 2017). This filtering procedure reduced the number of analyzed species to 828.
For this set of 828 genomes, we examined the correlation between the estimated number of ZNF and
retroelement ORFs. This analysis revealed a significant positive correlation across all metazoans, as well
as significant positive correlations across major phyla with at least 15 representative species, with the
exception of nematode worms (Fig. 2A). Within smaller taxonomic groups, this relationship holds in most
cases tested (Supp. Data 1), with some notable exceptions. For example, birds have lost most KZNFs
(Imbeault et al., 2017), such that the median number of ZNF ORFs in birds is a quarter that of other
chordates (121 vs 489); correspondingly, there is no correlation between ZNF and retroelement copy
number. Similarly, we observe no correlation across dipteran flies, which possess ZAD-ZNFs – these
have been studied in some depth in Drosophila melanogaster and have diverse roles in heterochromatin
organization, but do not appear to specifically target TEs for silencing (Kasinathan et al., 2020; Chung et
al., 2007; Baumgartner et al., 2022). Notwithstanding these exceptions, our analysis indicates that ZNF
and TE copy numbers are positively correlated across the breadth of metazoans.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint

8
Figure 2. ZNF copy number correlates with genomic TE content
A) Spearman’s rank correlations between estimated number of retroelement insertions and ZNF ORFs.
Each point is a representative species from a taxonomic family. Subplots on right hand side show
correlations for individual phyla. B) Uncorrected Spearman’s rank correlation between TE content and
FZNF copy number in cyprinid fish. C) Spearman’s rank correlation on phylogenetically independent
contrasts.
10
1
10
2
10
3
10
4
10
5
10
6
Retroelement copy number
10
0
10
1
10
2
10
3
ZnF copy number
ρ = 0.62
p = 3.1e-88
n = 828
10
2
10
4
10
6
10
0
10
1
10
2
10
3
ρ = 0.64
p = 6e-56
Chordata
n = 469
10
2
10
4
10
6
10
0
10
1
10
2
10
3
ρ = 0.50
p = 8e-15
Arthropoda
n = 212
10
2
10
4
10
6
10
0
10
1
10
2
10
3
ρ = 0.72
p = 5e-07
Mollusca
n = 37
10
2
10
4
10
6
10
0
10
1
10
2
10
3
ρ = -0.40
p = 3e-02
Nematoda
n = 30
20
25
30
35
40
45
50
55
TE content of genome (%)
0
100
200
300
400
500
600
700
Predicted FiNZ-ZnF genes
D. translucida
D. tinwini
P. tetrazona
C. idella
L. rohita
D. rerio
ρ = 0.650
−60
−40
−20
0
20
40
60
80
100
Contrast in TE content
−1000
−500
0
500
1000
1500
2000
Contrast in FiNZ-ZnF copy number
ρ = 0.542
p = 2.90e-03
B C
A
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
9
To further control for phylogenetic inter-dependence, we focused on the FZNF family in cyprinid fish,
making use of our refined gene annotations and improved estimates of genomic TE content generated
with dnaPipeTE (Goubert et al., 2015). Using these more accurate annotations, we find that the
correlation between ZNFs and TEs persists (Fig. 2B). Importantly, this relationship holds when explicitly
controlling for species relatedness by comparing phylogenetically independent contrasts in FZNF copy
number and TE content (Fig. 2C).
Studies of the relationship between KZNFs and TEs thus far have been retroelement-centric, since widely
used mammalian model organisms typically have relatively few DNA transposons (Platt et al., 2018). In
contrast, cyprinid fish have a wide diversity of different TE classes, and particularly DNA transposons
(Chang et al., 2022; Chen et al., 2019; Howe et al., 2013; Xu et al., 2014). We therefore asked whether
the correlation between cyprinid FZNFs and TEs held across different TE classes. It is not clear whether a
correlation with DNA transposons is expected, since these are typically less transcriptionally active than
retroelements, at least in zebrafish (Chang et al., 2022), and might not be targeted by ZNFs. Yet, we
found that the correlation was actually the strongest for DNA transposons (Supp. Fig. 4; rho = 0.6, p = 4e-
04, phylogenetically independent contrasts), and in the case of LINEs there was no significant correlation.
These results suggest that the relationship between FZNFs and TEs is not solely driven by
transcriptionally active retroelements but applies to a broad range of TEs.
There are two non-mutually exclusive explanations for the correlation between TEs and ZNFs. First,
ZNFs may physically interact with TEs, as is the case with KZNFs (Imbeault et al., 2017; Ecco et al.,
2017). Second, the TE content of the genome may have an enhancing effect on the rate of gene
duplication, such that genomes with large TE content will tend to have larger gene families. The latter
model is conceivable because TE accumulation is thought to promote ectopic recombination – and
therefore gene duplication or deletion – in a length- and copy number-dependent fashion (Charlesworth
and Langley, 1989; Tollis and Boissinot, 2012; Jurka et al., 2004). Under this latter model, one would
predict that TE content would positively correlate not only with the copy number of ZNFs but also that of
other expansive gene families. To explore this possibility, we used the same approach as used to count
ZNFs to estimate the copy number of mammalian olfactory receptors. Olfactory receptors are membrane
proteins known to turn over rapidly across species but are not expected to interact physically with TEs. In
a sample of representative species from 103 mammalian families, we observed a non-significant
correlation between the number of retroelement and olfactory receptor ORFs (Spearman’s rho = 0.12,
Supp. Fig. 5). While this analysis does not rule out the possibility that TE content has an impact on the
rate of gene duplication, it does imply that the effect is insufficient to explain the correlation observed
between ZNFs and TEs.
ZNFs are predicted to bind TE sequences
Based on our findings thus far, we reasoned that ZNF proteins are most likely interacting with TEs
through sequence-specific recognition of their DNA sequences (although occasionally ZNFs also bind
RNA (Font and Mackay, 2010)). We therefore sought to test whether ZNF binding motifs are enriched in
TE sequences. Defining the binding specificity of individual ZNF genes experimentally is labor-intensive,
but computational methods have been developed to predict ZNF binding motifs directly from their protein
sequence (Persikov and Singh, 2014; Najafabadi et al., 2015; Molparia et al., 2010; Kaplan et al., 2005).
The most recent of these methods predicts binding motifs for individual ZNF domains using a random
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint

10
forest classifier (Najafabadi et al., 2015). We used this approach to predict DNA binding motifs for all
ZNF ORFs from seven metazoan species with curated libraries of TE consensus sequences: four species
with a relatively large repertoire of poorly characterized ZNFs (octopus, zebrafish, rice weevil and sea
urchin), two “positive control” species known to use ZNFs to target TEs (human and mouse) , and an
expected “negative control” species, Drosophila melanogaster, whose ZNFs are apparently not targeting
TEs directly (Supp. Data 3).
First, we used previously published ChIP-exo data for human KZNFs (Imbeault et al., 2017) to confirm
that computationally predicted DNA binding motifs were similar to those experimentally determined.
Searching for each of 236 experimentally determined KZNF binding sites in the set of predicted binding
motifs, we found that 11 (5%) produced significant matches to their predicted counterpart (q-value <
0.05), compared to 0 matches after shuffling the predicted motifs. Similar results have been demonstrated
by others working with mice (Wolf et al., 2020). As a second positive control, we compared the predicted
binding motifs for human and mouse ZNF ORFs to libraries of their consensus TE sequences and
observed a significant enrichment of predicted matches within TEs, compared to matches using shuffled
motifs (Fig. 3A). This result is expected given that KZNFs are known to target TEs and confirms that
predicted binding sites recapitulate, at least in part, experimentally obtained motifs. For example, we
were able to recapitulate human ZNF320 binding to LTR14a and ZNF483 binding to L1PA7 (Supp. Fig.
6). These controls demonstrate that while computational prediction of ZNF binding specificity is far from
solved, it is sufficient to capture a proportion of biologically relevant binding activity.
Figure 3. ZNFs are predicted to bind
TE sequences. A) Percentage of
sequences matched by predicted ZNF
motifs, with a q-value cutoff of 0.05.
Grey bars represent matches to
libraries of consensus TE sequences,
while blue bars represent matches in
equally sized libraries of non-
repetitive genomic sequence. Green
bars represent shuffled motifs,
searched against consensus TE
libraries. P-values are calculated as
one-tailed binomial tests for the
probability of observing at least x
matches in the TE library, using
binding frequency to non-repetitive
sequences or with shuffled motifs as a
base probability. B) Selected matches
between Danio rerio consensus TE
sequences and ZNF ORFs.
We therefore turned our attention to the four species whose ZNFs are less studied (zebrafish, octopus, rice
weevil, sea urchin), predicting binding motifs for all ZNF ORFs within each species and examining the
Copia-11
q = 5e-04
hAT-N102B
q = 1e-03
L1-26
q = 1e-03
Copia-13E
q = 7e-03
Merlin-2
q = 6e-03
DIRS-8
q = 2e-02
Zisupton-6
q = 2e-02
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
p ≈ 0.0
A B
0 20 40 60 80
Sequences targeted (%)
H. sapiens
M. musculus
D. rerio
O. sinensis
D. melanogaster
S. oryzae
S. purpuratus
p = 6.8e-07
520
p = 1.1e-18
448
p = 2.6e-183
2286
2882
n.s.
685
p = 2.5e-14
3393
3305
p = 4.7e-25
p = 3.9e-23
p ≈ 0
p = 5.1e-48
p = 1.0e-23
p = 1.3e-162
TE sequence
Non-repetitive
Shuffled
p ≈ 0
p ≈ 0
p ≈ 0
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
11
frequency of these motifs in consensus TE sequences derived from each respective species (Fig 3A).
Using a false discovery threshold of q = 0.05, we searched for significant matches between our predicted
ZNF binding motifs and TE consensus sequences, then compared this to equivalently sized libraries of
non-repetitive genomic sequence. For all four species, we observed a significant enrichment of matches to
the TE sequences compared to non-repetitive sequences (Fig 3A). By contrast, we do not observe such
enrichment for Drosophila melanogaster. Rather, for that species we found a greater percentage of non-
repetitive sequences matched by predicted ZNF binding motifs than expected from shuffled motifs (Fig
3A). This finding is consistent with previous functional studies of Drosophila ZAD-ZNFs (Kasinathan et
al., 2020) and our observation that the copy number of dipteran ZNFs does not correlate with the number
of TE copies (Supp. Data 1). Notably, we also found that in all species both retroelements and DNA
transposons were predicted to be targeted by ZNFs (Supp. Data 3).
As an additional control, we repeated the analysis using shuffled motifs, and found in all species that the
predicted motifs bound a higher percentage of TE sequences compared to the shuffled motifs (Fig. 3A).
We note, however, that shuffled motifs still recognize a substantial fraction of sequences in most species,
likely due to the fact that ZNFs are often predicted to bind simple sequence repeats or homopolymeric
tracts that would be unaffected by shuffling (e.g. Fig. 3B). Finally, to test species specificity, we searched
the Danio rerio TE library using human ZNFs and compared these to zebrafish ZNFs. We found that
zebrafish ZNFs were far more likely to target zebrafish TEs than were human ZNFs (odds ratio = 29.5, p
≈ 0.0, Fisher’s exact test). These results suggest that the preferred binding of ZNFs to TE sequences is not
unique to mammalian KZNFs but likely a general property of many metaozan ZNF families.
FZNFs are under positive selection in zebrafish
TEs evolve rapidly and are under strong selection to evade repression by their hosts; many KZNF proteins
are in direct competition with TE families, leading to signatures of arms races between the two (Bruno et
al., 2019; Fernandes et al., 2018; Jacobs et al., 2014). One such signature is positive selection (i.e.
adaptive evolution) acting on the DNA contacting residues of ZNFs, i.e. those involved in TE recognition
(Emerson and Thomas, 2009). We therefore tested for evidence of positive selection in Danio rerio
FZNFs by comparing rates of synonymous and non-synonymous substitutions (dN/dS). Values of
dN/dS > 1 denote an excess of non-synonymous changes and suggest that natural selection accepts novel
amino-acid changes at a rate faster than expected under a neutral model.
In practice, it is challenging to confidently align ZNFs due to their repetitive nature and tendency to
undergo internal rearrangements and gene conversion, and errors in this process can lead to false positives
in assessment of dN/dS ratios (Mallick et al., 2009). To account for this, we restricted our analyses to
seven clades of recently duplicated paralogs unique to Danio rerio (with a minimum of ten members - see
methods for details). Using PAML (Yang, 2007), we performed likelihood ratio tests to compare two
models of evolution for each clade – one in which the genes were evolving under purifying selection, and
one in which some sites were assumed to be under positive selection (M2 vs M2a). In four of these seven
clades, the model featuring positive selection (M2a) was significantly favored (p-value < 0.01, likelihood
ratio tests, Supp. Data 4). Furthermore, in two clades we found that base-contacting residues of ZNF
domains were significantly enriched for values of dN/dS > 1 (odds ratio = 2.45, p-value = 0.01; odds ratio
= 2.84, p-value = 0.02; Fisher’s exact test).
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
12
As an alternative to dN/dS analyses, which are dependent on accurate gene alignments, we calculated the
sequence entropy at positions in the canonical ZNF domain for each species in Fig. 3. The results
revealed a clear trend for base-contacting residues to be amongst the most variable in sequence, with the
possible exception of Drosophila melanogaster (Supp. Fig. 7). These data, combined with the enrichment
of ZNF predicted binding sites in TE sequences, suggest that zebrafish ZNFs, and likely those of many
other species, are evolving adaptively to target specific TE families.
FZNFs are expressed in distinct waves during embryogenesis
TEs – retroelements especially – are highly active during metazoan embryogenesis, as this provides them
with the opportunity to transpose in the cells giving rise to the adult germline, thus ensuring the vertical
inheritance of new insertions (Haig, 2016; Ansaloni et al., 2019; Rodriguez-Terrones and Torres-Padilla,
2018; Chang et al., 2022). Previous work has shown that zebrafish ZNFs are expressed at the onset of
zygotic genome activation (ZGA), mirroring the pattern of KZNFs in human and mice, and consistent
with a potential role in TE silencing (Pontis et al., 2019; White et al., 2017; Hadzhiev et al., 2019). We
sought to further explore the timing of FZNF expression, predicting that if they are deployed to repress
TEs, their expression should overlap with that of TEs. Using our de-novo FZNF annotations, we
remapped previously published RNA-sequencing data covering the first 24 hours of zebrafish
development (White et al., 2017). Setting a lower limit of 0.5 transcripts per million (TPM) to call genes
as expressed, we find that approximately half of all FZNFs (306 out of 684) are expressed during early
development.
Recently published work identified two distinct waves of ZNF expression in zebrafish, named “sharp
peak” and “broad peak” (Hadzhiev et al., 2019; Hadzhiev et al., 2021). Sharp peak FZNFs share a distinct
promoter architecture consisting of a clearly identifiable TATA-box and are expressed in the minor wave
of ZGA, followed shortly after by TATA-less broad peak ZNFs in the major wave. With our updated
FZNF annotations, we recapitulated these findings, observing expression of 80 sharp peak and 204 broad
peak genes, hereafter termed “dome”, and “late” based on the timing of their expression (Fig. 4A). The
timing of these peaks overlaps with that of most TE families, and in the case of dome stage FZNFs,
precedes almost all zygotic TE transcription.
We also found a small subset of approximately 22 FZNF genes with maternally deposited transcripts (Fig.
4A). To our knowledge, these have not previously been described, likely due to their incomplete
annotation in Ensembl and RefSeq gene sets. Furthermore, maternally-deposited mRNAs in zebrafish and
other animals are, initially, only partially poly-adenylated (Winata et al., 2018; Cui et al., 2013; Potireddy
et al., 2006), and are therefore difficult to detect when using polyA-selected RNA-seq platforms. As the
White et al. data falls into this category, we remapped rRNA-depleted reads from a second dataset
covering early embryogenesis (Winata et al., 2018), which confirmed that these maternally-deposited
transcripts are indeed present in both the egg and 1-cell zygote (Fig 4B).
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint

13
Figure 4. Embryonic expression of FZNF genes in zebrafish
A) Median expression trajectory of 306 genes with expression of at least 0.5 TPM in at least 1 stage,
separated by cluster. Shaded borders represent 90% confidence intervals. Light grey bar shows
approximate timing of ZGA. Expression trajectory of maternally deposited FZNFs prior to 1000-cell
stage is dashed to reflect the fact that poly-A selected RNA sequencing is not an accurate reflection of
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
14
true mRNA levels prior to ZGA. B) FZNF expression from rRNA-depleted reads, prior to ZGA. C)
Maternally deposited genes are significantly older, whilst those expressed specifically at Dome stage are
young, and generally unique to D. rerio. Ages were quantified as branch lengths on a neighbor-joining
phylogenetic tree generated from pairwise distances. D) The majority of maternally deposited FiNZ genes
are located in a specific, sub-telomeric cluster. Each vertical line represents the midpoint of an expressed
FZNF gene.
Given the markedly different expression pattern of maternally deposited FZNFs, we looked for other
features differentiating them from their zygotically expressed counterparts. While the majority of Danio
rerio FZNFs are recently duplicated, particularly dome stage genes, those that are maternally deposited
are conserved across species and are significantly older (Fig. 4C, Supp, Data. 5). Moreover, they are
physically co-localized in the subtelomeric region of Chr 4q (Fig. 4D), which lacks the repeat density that
characterizes much of Chr 4q (Chang et al., 2022). The age of maternally deposited genes, as well as their
degree of species conservation, is hard to reconcile with a role in TE targeting, as TEs turn over rapidly
and are frequently species-specific. Rather, the deeper conservation and physical clustering of these
maternally deposited FZNFs is suggestive of a role in development that differs from that of Dome and
Late-stage FZNFs.
FZNFs repress LTR retroelement expression during early development
Next, we turned to the function of FZNF genes. Based on their embryonic expression, predicted binding
specificity and strong correlation with TE content, we predicted that the bulk of FNZF genes play a role
in TE silencing. If true, then knocking down their expression ought to lead to a corresponding increase in
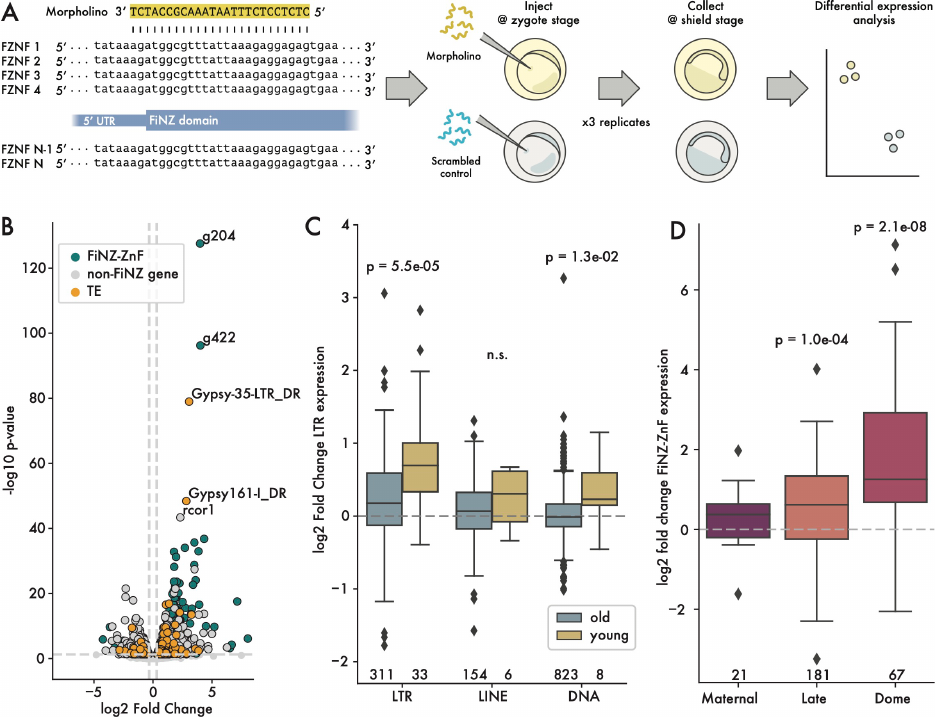
TE expression. To test this, we employed a Morpholino oligonucleotide strategy aimed at blocking
translation of most FZNF proteins simultaneously. By aligning the first exon of the FZNF annotations
from zebrafish, we were able to design a Morpholino predicted to target the translation start site of 447
out of 684 annotated FZNFs (Fig. 5A). We injected zebrafish embryos either with this Morpholino or a
scrambled control, then compared gene and TE expression using RNA-seq. Embryos were collected for
sequencing at shield stage, as this corresponds with a period at which many TE families and most FZNFs
are robustly expressed (Fig. 4A).
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
15
Figure 5. Up-regulation of LTR retroelements in response to FZNF knock down
A) Cartoon showing design of Morpholino-based, translation-blocking FZNF knock-down experiment. B)
Log2 fold change vs p. value, comparing FiNZ translation blocking morpholinos to scrambled
morpholino control. C) Log2 fold change comparison between old and young TE families, with “young”
being defined as those whose median insertion age was less than 0.01 substitutions per site. P-values are
Wilcoxon rank-sum tests comparing old and young in each TE class. D) Log2 fold change of different
FZNF categories. P-values are binomial tests for each category, assessing likelihood of seeing at least x
upregulated genes, assuming a null hypothesis where there is a 50% chance of being either up- or down-
regulated. Numbers below boxes in C) and D) give number of data points per box.
After sequencing and read mapping, we carried out quality controls before comparing TE expression
between treatment and control groups. First, we performed principal component analyses of batch-
corrected samples, which showed clear separation between the FZNF knock-down and control groups
(Supp. Fig. 8A). Using a false discovery rate threshold of 0.05 and an absolute log
2
fold change of at least
0.32 to call differentially expressed genes and TE families, we observed a total of 999 differentially
expressed genes, excluding FZNFs and TEs (Supp. Data 6). Amongst these, a GO-term enrichment
analysis revealed no notable enrichment for biological processes beyond general terms related to
embryonic development, metabolic processes and translation (Supp. Data 7). Importantly, we did not
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
16
observe any enrichment for stress response pathways, nor did we observe a significant change in tp53
levels (log
2
fold change = -0.01), which has been known to cause off-target effects in Morpholino knock-
downs (Bedell et al., 2011). Overall, there was no significant difference in the number of up- vs down-
regulated non-FZNF genes (p = 0.02, binomial test).
As the expression of TEs and FZNF genes is highly dynamic during the first 24 hours of zebrafish
development (Chang et al., 2022; Laue et al., 2019; Guo et al., 2021), we sought to ensure that the
embryos were staged appropriately, as differences in the relative stage of FZNF knockdown and control
groups could lead to artefactual observations of differential expression. We therefore compared our
samples with samples from White et al., covering 50% epiboly, shield stage, and 75% epiboly (White et
al., 2017). Treating these samples as additional control groups, and after batch correction, we found that
our samples clearly clustered with shield stage samples from White et al. (Supp. Fig. 8B, C).
We then asked whether TE expression was up-regulated in the FZNF knock-down group. Looking at the
overall number of differentially expressed TE families, we found that TEs were significantly more likely
to be up-regulated than down-regulated, consistent with de-repression following FZNF knockdown (odds
ratio = 3.78, p-value = 6.1e-08, Fisher’s exact test; Fig. 5B, Supp. Fig. 8D-E). Separating by TE class, we
found that this trend was driven predominantly by LTR elements (odds ratio = 6.56, p-value = 1.2e-08,
Fisher’s exact test; Supp. Fig. 9A). Since LTRs include many of the youngest TE families in the zebrafish
genome (Chang et al., 2022), we reasoned that their enrichment amongst up-regulated genes and TE
families may reflect a tendency for young, active families to be most intensively targeted by FZNFs. We
therefore compared young and old TE families to each other, defining the former as those whose
insertions averaged fewer than 0.01 substitutions per site (Chang et al., 2022). This analysis revealed that
the magnitude of de-repression following FiNZ-knockdown was significantly greater amongst young
families (p = 5.5e-05, Wilcoxon rank sum test), and that this trend held across different TE classes (Fig.
5C). Thus, FZNF knockdown leads to significant de-repression of young, potentially active TE families.
In addition to age, we also looked at the timing of expression, since by collecting at “shield” stage
(approximately 6 hours post fertilization), we would not be able to observe the effects on those TEs
whose expression peaked at later time points. Using hierarchical clustering to separate TEs into groups
with expression peaks either before or after shield stage, we found that those TE families which were
expressed earlier were significantly more up-regulated than those expressed later (Supp. Fig. 9B).
Curiously, the genes most up-regulated by FZNF knockdown were FZNFs themselves, for example, g204
and g422 (Fig. 5D). While we cannot currently rule out the possibility that the Morpholinos inhibit the
degradation of their target sequences, to our knowledge, such effects have not previously been reported in
the literature. Alternatively, these results suggest that FZNF genes are themselves targeted for repression
by FZNF proteins, thus forming a negative feedback loop. In this way, their expression would be robustly
switched off once sufficient FZNF protein had been generated, without the need for additional regulatory
control. Such negative feedback loops have recently been documented for mammalian KZNFs (Pontis et
al., 2022).
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
17
Discussion
In this work, we observed a deep coevolutionary relationship between TEs and ZNFs that spans the
breadth of metazoans. This is most clearly seen in the strong positive correlation between TE and ZNF
copy numbers, which persists across at least 750 million years of animal evolution and is independent of
the various accessory domains that characterize different ZNF families. We also found that ZNFs from a
range of metazoans are predicted to bind TE sequences, and in the case of FZNFs in zebrafish, are
evolving under positive selection in ZNF arrays. Finally, through simultaneous knock-down of a large
subset of FZNFs during zebrafish embryogenesis, we demonstrated that this family represses young,
recently active TEs. Since the FiNZ domain is unique to cyprinid fish, which lack both the KRAB domain
and its cofactor KAP1 (a.k.a. TRIM28), the silencing abilities of FZNFs and KZNFs appear to have
evolved convergently. This suggests that the role of ZNFs in TE targeting and silencing is driven by the
intrinsic plasticity of ZNF DNA binding activity, as opposed to being a function of specific accessory
domains.
The extensively duplicated ZNF families of metazoan species are rarely observed in other eukaryotic
taxa, where instead, small-RNAs acting in concert with Argonaute proteins are the primary drivers of TE
heterochromatinization (Hall et al., 2002; Volpe et al., 2002; Allshire and Madhani, 2018; Heger et al.,
2020; Najafabadi et al., 2017). Thus, ZNF-based TE targeting is likely to be a metazoan innovation.
However, there are several metazoan groups whose ZNF families appear to behave atypically. For
example, we found no correlation between ZNFs and retroelements in dipteran flies, which includes
intensively studied species such as Drosophila melanogaster and Anopheles gambiae, nor did we observe
a binding preference for TEs in Drosophila ZNFs. However, in Drosophila the piRNA system (itself a
branch of the Argonaute-siRNA system) is directly involved in pre-transcriptional silencing of TEs during
embryogenesis, with piwi knockdowns resulting in a failure of H3K9me3 to establish over otherwise
silenced TE loci (Sienski et al., 2012; Wei et al., 2021; Fabry et al., 2021). Less is known about
mosquitoes, but it is clear that they have undergone numerous duplications of piwi homologues, and have
a complex suite of piRNAs that includes many arising from non-TE satellite sequences (Ma et al., 2021;
Gamez et al., 2020). Similarly, nematode worms, which typically have few ZNF genes, have undergone
extensive diversification of Argonaute genes (Buck and Blaxter, 2013). While research into the function
of these Argonaute homologs remains in the early stages (Seroussi et al., 2022), both the dipteran and
nematode cases point to potential functional overlap between the Argonaute/piRNA system and ZNFs.
It is still an open question whether silencing of TE transcription per se, is the primary function of
metazoan ZNFs as a form of genome defense system. While depletion of both KZNFs and FZNFs (this
study) have now been shown to unleash TE expression, there are many paradoxical observations that
challenge the idea of ZNF as an adaptive defense system against TE invasion and suggest transcriptional
repression itself may be secondary to other functions. First and foremost, it appears that ZNFs evolve at a
glacial pace relative to the speed at which novel TE families can establish and propagate within a species.
For example, the DNA transposon P-element spread throughout all wild populations of Drosophila
melanogaster in the space of approximately 30 years, and the retrovirus KoRV has been spreading in
koalas exogenously and endogenously over a similar period (Anxolabéhère et al., 1985; Tarlinton et al.,
2006); in contrast, observations in primates imply that the emergence of novel KZNF repressors takes
place over millions of years (Imbeault et al., 2017; Ecco et al., 2017). On the other hand, the piRNA
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
18
system is able to adapt much more rapidly, as TE insertions themselves form the raw material for novel
piRNAs – indeed, in the case of both KoRV and P-element, piRNAs have already emerged to mitigate
their deleterious effects (Brennecke et al., 2008; Yu et al., 2019). Thus, with respect to their ability to
quickly respond to novel TEs, the piRNA system appears better suited as a front-line defense than ZNFs.
In addition to the speed at which they evolve, other features of ZNFs are also difficult to reconcile with a
general function in repressing TE transcription. While the majority of KZNFs are expressed during early
embryogenesis and specifically target TEs, approximately a third target non-TE sequences, such as
satellite repeats, gene promoters, and even other KZNFs (Imbeault et al., 2017). There are also examples
in both mouse and human of conserved clusters of KZNFs that target TEs younger than themselves,
implying they had targets predating the emergence of their current TE families (Imbeault et al., 2017;
Wolf et al., 2020; Wolf et al., 2015). These observations have led others to propose that, rather than
simply repressing TEs, KZNFs play an important role in their domestication by taming their cis-
regulatory activities and facilitating their integration into existing gene regulatory networks(Ecco et al.,
2017; Pontis et al., 2019; Bruno et al., 2019). Under this model, KZNFs would act as tolerogenic agents
promoting the evolution of species-specific gene regulatory networks. While this is an attractive model, it
is important to clarify that this feature of KZNFs cannot be the initial driver of their rapid evolution and
turnover, since TE domestication is a process that takes place over many generations, whereas selection
primarily acts on currently extant individuals (Sniegowski and Murphy, 2006).
We therefore propose a non-mutually exclusive alternative to the transcriptional repressor hypothesis, in
which metazoan ZNFs instead serve as genome stabilizers, protecting DNA from ectopic recombination
by nucleating heterochromatin to repetitive regions of the genome. Heterochromatin has a strong
suppressive effect on homologous recombination and prevents the formation of gross chromosomal
rearrangements (Allshire and Madhani, 2018; Grewal, 2010). For example, histone H3K9 methylation
prevents repeat- and/or TE-mediated recombination and replication stress in worms and fission yeast
(Zeller et al., 2016; Sasaki et al., 2010). Importantly, the propensity of TEs to cause deleterious
chromosomal rearrangements via non allelic recombination is thought to be the primary factor preventing
their accumulation in genomes (Allshire and Madhani, 2018; Sasaki et al., 2010; Kent et al., 2017; Tollis
and Boissinot, 2012). Thus, the deployment of sequence-specific ZNFs to deposit heterochromatin over
discrete TE families might explain in part why some organisms can sustain large-scale amplification of
TE families as seen in a subset of animal genomes such as octopus, weevils, sea urchin, cyprinid fish, and
mammals to name a few highlighted in this study. If genome stabilization is the principal function of most
metazoan ZNFs, then it relieves the requirement for rapid adaptation to novel TEs, since the effect of TEs
on rates of non-allelic homologous recombination are not felt until the TEs in question have reached
significant copy number. Similarly, it would explain why satellite sequences or non-autonomous DNA
elements, which do not need to be transcribed to amplify, appear to be targets of many ZNFs.
Much remains to be discovered about the role of ZNF genes in metazoan evolution, but with the wealth of
high-quality genome assemblies and improved tools for manipulating gene expression in a variety of
organisms, it is now possible to answer many of the long-standing questions about this extraordinary gene
family. In this work, we have demonstrated that the co-evolutionary relationship between TEs and ZNFs
extends well beyond KZNFs and is likely a metazoan innovation for TE silencing and/or stabilization. In
zebrafish, FZNFs have independently expanded to repress young, recently active TE families during early
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
19
embryonic development. Across animals, there are dozens of other such uncharacterized ZNF families,
many of which may reveal exciting new biology, and which have potential uses in the development of
genome engineering tools.
Methods
Predicting ZNF ORF copy number
Initial estimates of ZNF ORF copy number were achieved by extracting all possible ORFs from 3,221
representative metazoan genomes available from the NCBI assembly database, as of 2021-12-06. To
facilitate this process, we set minimum and maximum ORF sizes of 375 and 10,000 base pairs,
respectively. The resulting amino-acid sequences were searched for potential C2H2 ZNF domains using
hmmsearch command line tool from the HMMER suite (Eddy, 2011), and the PFAM “zf_C2H2” (PFAM:
PF00096) HMM as a query profile (Mistry et al., 2021). Resulting hits were merged if they were within
100 residues of each other, and filtered to exclude sequences with fewer than five ZNF domains in total.
Annotation of FZNF genes
For improved annotations of FZNF genes in 32 cypriniform fish and a milkfish outgroup, we first
identified genomic regions containing candidate FZNF genes using BLAST to search for matches with a
consensus FiNZ domain sequence. This consensus sequence was generated from a set of D. rerio FZNF
genes previously identified as being expressed during development (White et al., 2017). Using the “PPX”
module of Augustus v3.3 (Hoff and Stanke, 2018; Keller et al., 2011), we carried out a first pass of the
genomic regions identified in our set of cypriniform species, using a protein profile generated from the
previously mentioned set of D. rerio annotations. To avoid biasing our search towards zebrafish, we used
the results of this round of annotation to generate a new FZNF profile generated by sampling genes from
all species. We then repeated the above procedure, and performed a final filtering step with HMMER
(Eddy, 2011) to identify annotated genes containing both the FiNZ domain and ZNF domain (PFAM:
PF00096).
For D. rerio specifically, we produced a separate, high-quality set of gene predictions by retraining
Augustus specifically for FZNFs using a manually curated set of Ensembl predictions. This allowed us to
generate predictions for 5’ and 3’ untranslated regions, for use downstream in analyses of gene
expression. We explored the effect of including transcripts generated by Trinity(Grabherr et al., 2011) as
hints for Augustus, but found that these reduced the quality of resulting annotations, likely as a result of
the difficulty of assembling accurate transcripts for such repetitive genes. Full parameter details are
available online.
Predicting genomic TE content
We used two approaches to estimate genomic TE content. In the first, we focused on retroelements, using
an identical approach to that which we used to estimate ZNF ORF number. In place of the PFAM
zf_C2H2 domain, we instead used a selection of reverse transcriptase and RNaseH profiles (PF00078,
PF07727, PF13456, PF00075, PF17917, PF17919). Resulting hits were merged as done for ZNFs, but no
restrictions were placed on the number of hits per ORF. For more careful estimates of TE content for use
when focusing on FZNF genes, we estimated the TE-derived proportion of genomes using
dnaPipeTE(Goubert et al., 2015), a tool that offers considerable speed improvements over
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
20
RepeatMasker/Modeller with comparable accuracy. This program requires short reads and genome size as
input: for the former, we simulated reads using ART(Huang et al., 2012) and for the latter, we used
genome assembly size as a proxy for true genome size.
Cypriniformes phylogeny
To generate a phylogenetic tree of the Cypriniformes order (required to account for phylogenetic non-
independence in comparative genomic analyses) we first used the Actinopterygii-specific BUSCO
database to extract intact single-copy orthologues from our set of 33 species. We then selected those
protein sequences found in at least 10 out those species, producing a final set of 3,581 proteins. These
were aligned separately using mafft v7.475 with parameters –globalpair and –maxiterate 1000 (i.e. ginsi)
(Katoh and Standley, 2013) and trimmed to remove large insertions or poorly aligned regions using
trimAl v1.4.rev15 with the –automated1 parameter set (Capella-Gutiérrez et al., 2009). All resulting files
were combined to produce a concatenated super-gene alignment, which was used as input when
generating a time-calibrated phylogeny.
IQ-TREE v2.0.6 was used to generate a maximum likelihood tree from a partitioned analysis of the super-
gene alignment (Minh et al., 2020; Chernomor et al., 2016; Hoang et al., 2018). Ultrafast bootstraps and
Shimodaira–Hasegawa approximate likelihood ratio tests were used to determine branch support values,
with 5000 and 1000 replicates respectively. A time-calibrated tree was generated using the integrated
LSD2 module (To et al., 2016), constraining the date of the split between Gonorynchiformes and
Cypriniformes (i.e. between Chanos chanos and all other species) to 162 Mya, based on a recent
comprehensive phylogeny of teleost fish (Hughes et al., 2018).
Predicting ZNF binding sites
To predict ZNF binding sites, we made use of the previously published tool, ZiFRC, with ZNF ORFs
identified from our initial genome searches as input (Najafabadi et al., 2017; Najafabadi et al., 2015). To
test for enrichment of predicted ZNF binding sites in various sequence sets, we used fimo and tomtom
from the MEME suite of motif enrichment tools (v5.3.3) (Bailey et al., 2015). First, tomtom was used to
compare our predicted motifs to motifs obtained from human ChIP-seq datasets (Imbeault et al., 2017).
Following this positive control, we used fimo to calculate motif enrichment in three separate cases for
each species.
In the first case, we searched our library of predicted ZNF binding sites against libraries of TE consensus
sequences. For human, mouse, fruit fly and zebrafish, these libraries were taken directly from
RepBase26.10, in the case of sea urchin and rice weevil, they were provided by direct communication
with the authors of previously published studies on the TE content of these species (Panyushev et al.,
2021; Parisot et al., 2021), and in the case of octopus, we generated a TE consensus sequence library
using RepeatModeller v2.0.1 (Flynn et al., 2019).
For the second case, we searched a library of randomly selected non-repetitive genomic sequences,
selected using bedtools v2.30.0 (Quinlan and Hall, 2010); this was done in a manner ensuring that, for
each species, the resulting library size and distribution of sequence lengths was identical to that of the TE
sequence library. In the third case, we searched the TE consensus sequences again, this time using
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
21
shuffled binding motifs. Finally, we performed a cross-species comparison between zebrafish and human
using fimo.
Testing for positive selection
To test for evidence of positive selection in FZNF genes, we first searched for recently duplicated in-
paralogs. As multiple sequence alignments of ZNF genes can be unreliable, we calculated pairwise
Needleman-Wunsch alignments between all pairs of 6,732 annotated genes from 29 cypriniform species
with at least one FZNF gene, using the “needle” command line tool from the EMBOSS suite, v6.6.0 (Rice
et al., 2000). The resulting pairwise distance matrix was used as input to generate a neighbor-joining
phylogenetic tree, using the EMBOSS “fneighbor” tool. This gene tree was used to identify clades of
recently duplicated in-paralogs within the cypriniform species tree. We selected seven clades from Danio
rerio, where each clade contained at least ten members, and all members were within 30 amino-acids of
each other in length. Each one of these clades was realigned with Prank, using a codon substitution matrix
(Löytynoja, 2014; Kosiol et al., 2007), and all gaps in the resulting alignment were removed using using
trimAl v1.4.rev15 (Capella-Gutiérrez et al., 2009). Finally, guide trees were generated for each clade
using IQ-TREE v2.0.6 with a codon substitution model (Minh et al., 2020).
To test for evidence of positive selection, we used the “codeml” tool from the PAML4 software suite
(Yang, 2007). We compared two sets of nested site models: first, M1a (NearlyNeutral) and M2a
(PositiveSelection), and second, M7 (beta) and M8 (beta & ω). The transition to transversion ratio, κ, was
calculated from the guide tree, while the dN/dS rate ratio, ω (i.e. the parameter being tested) was
estimated from the data. Likelihood ratio tests with two degrees of freedom were used to compare M1a to
M2a, and M7 to M8, respectively.
Reanalysis of zebrafish developmental transcriptome data
To assess FZNF gene and TE expression, we used first used STAR (v2.7.5a) (Dobin et al., 2013) in multi-
mapping mode to re-align reads from White et al. (White et al., 2017) to the Danio rerio GRCz11
reference genome, setting the maximum intron/read pair gap size at 500,000. To count reads from genes
and TEs simultaneously, we used TEtranscripts-2.2.1’s TEcount tool in multi-mapper mode, with a GTF
file combining Ensembl gene annotations with our FZNF annotations, along with TE locations as
provided in Chang et al. (Chang et al., 2022). We used GTFtools v0.8.0 (Li et al., 2022) to calculate the
median length of each gene, and used these measurements to calculate TPM for each gene/TE, allowing
comparison of expression across stages. Specifically, for each transcript we calculate the sum of the union
of exons. For genes, we then take the median length of transcripts associated with each gene, whereas for
TEs we use the sum of all insertions in the genome, thus partially controlling for insertion copy number.
Morpholino knockdown of FZNF translation
We ordered a translation blocking Morpholino with the sequence
“CTCTCCTCTTTAATAAACGCCATCT” from Gene Tools LLC, which was sufficient to target 447 of
our predicted FZNF mRNA transcripts with no mismatches. Wild-type, Tübingen strain zebrafish were
maintained at 28°C on a 14/10hr light/dark schedule. Individual breeding pairs of male and female fish
were separated overnight to induce naturally synchronized spawning and fertilization the following
morning. To knock down FZNF expression, we injected 4.5ng of the custom Morpholino into fertilized
Danio rerio zygotes, and in parallel, we injected embryos with the same quantity of a randomized control,
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint

22
consisting of a mixture of up to 4
25
possible sequences. Embryos were collected at shield stage, and stored
at -20°C immediately after collection. Each pair of injections was repeated in triplicate, such that 23
treatment and 19 control embryos were in the first batch, 30 treatment and 30 control in the second, and
31 and 30, respectively, in the third. Total RNA extraction was performed using the Qiagen RNeasy kit.
RNA sequencing
We outsourced cDNA library preparation and sequencing to Novogene Inc. Samples were shipped in dry
ice, and after quality controls, strand-specific libraries were prepared using poly-A enrichment. These
libraries were pooled and sequenced in 150bp paired-end mode using the NovaSeq 6000 platform. After
receiving sequencing data, we carried out quality assessment using FastQC (v0.11.9) (Andrews, 2010),
and then mapped and counted reads according to the protocol previously described for reanalysis of
White et al. data. Following this, differential expression analyses were carried out using the DESeq2 R
package v1.26.0 (Love et al., 2014).
Code and data availability
All scripts used in the generation and analysis of data for this project can be found at GitHub. Code
relating to the broader analyses of metazoan ZNFs can be found at: https://github.com/jonathan-
wells/metazoan-znfs, while code relating to FZNFs specifically can be found at:
https://github.com/jonathan-wells/finz-znf. Supplementary data files containing processed data are
provided, and all raw data is available upon request. Sequencing reads generated through this work will
be available at NCBI SRA.
Acknowledgements
We thank Dr. Joseph Fetcho for zebrafish husbandry support. We thank members of the Feschotte
laboratory for valuable feedback and discussion. This work was supported by grant R35-GM122550 from
the National Institutes of Health to C.F. J.W. is supported by a Human Frontier Science Program long-
term fellowship (LT000017/2019-L). N.-C.C. is supported by a Distinguished Scholar Award from the
Cornell Center for Vertebrate Genomics. C.C. was supported by a stipend from the Cornell Research
Experience for Undergraduates program.
Author contributions
J.W. performed all computational analyses. N.-C.C. performed FZNF Morpholino injections. J.M. carried
out early studies of the FiNZ domain and provided general assistance with “wet-lab” experiments. C.C.
generated and analyzed preliminary data from surveys of metazoan ZNF and TEs. N.R. carried out early
annotation of Danio genus TEs and performed pilot wet-lab experiments. B.J. generated and analyzed
preliminary data for testing correlation between TEs and other gene families. J.W. designed all
experiments with input from N.-C.C., J.M. and C.F. J.W. wrote the manuscript and created figures with
input from C.F. Initial project conception by J.W., J.M. and C.F.
References
Albertin CB, Simakov O, Mitros T, Wang ZY, Pungor JR, Edsinger-Gonzales E, Brenner S, Ragsdale
CW, and Rokhsar DS. 2015. The octopus genome and the evolution of cephalopod neural and
morphological novelties. Nature 524: 220-24.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
23
Allshire Robin C and Madhani Hiten D. 2018. Ten principles of heterochromatin formation and function.
Nature reviews Molecular cell biology 19: 229.
Almeida MV, Vernaz G, Putman ALK, and Miska EA. 2022. Taming transposable elements in
vertebrates: from epigenetic silencing to domestication. Trends Genet 38: 529-53.
Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ. 1990. Basic local alignment search tool. J
Mol Biol 215: 403-10.
Andrews S. 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online].
Ansaloni F, Scarpato M, Di Schiavi E, Gustincich S, and Sanges R. 2019. Exploratory analysis of
transposable elements expression in the C. elegans early embryo. BMC Bioinformatics 20: 484.
Anxolabéhère Dominique, Nouaud Danielle, Périquet Georges, and Tchen Paul. 1985. <i>P</i> -element
distribution in Eurasian populations of <i>Drosophila melanogaster</i> : A genetic and molecular
analysis. Proceedings of the National Academy of Sciences 82: 5418-22.
Aravin AA, Hannon GJ, and Brennecke J. 2007. The Piwi-piRNA pathway provides an adaptive defense
in the transposon arms race. Science 318: 761-64.
Bailey TL, Johnson J, Grant CE, and Noble WS. 2015. The MEME Suite. Nucleic Acids Res 43: W39-49.
Baumgartner Lisa, Handler Dominik, Platzer Sebastian, Duchek Peter, and Brennecke Julius. 2022. The
Drosophila ZAD zinc finger protein Kipferl guides Rhino to piRNA clusters. bioRxiv
Bedell VM, Westcot SE, and Ekker SC. 2011. Lessons from morpholino-based screening in zebrafish.
Brief Funct Genomics 10: 181-88.
Bellefroid E. J., Poncelet D. A., Lecocq P. J., Revelant O., and Martial J. A. 1991. The evolutionarily
conserved Kruppel-associated box domain defines a subfamily of eukaryotic multifingered proteins.
Proceedings of the National Academy of Sciences 88: 3608-12.
Brennecke J., Malone C. D., Aravin A. A., Sachidanandam R., Stark A., and Hannon G. J. 2008. An
Epigenetic Role for Maternally Inherited piRNAs in Transposon Silencing. Science 322: 1387-92.
Bruno Melania, Mahgoub Mohamed, and Macfarlan Todd S. 2019. The Arms Race Between KRAB–Zinc
Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Annual Review of
Genetics 53: annurev-genet.
Buck Amy H. and Blaxter Mark. 2013. Functional diversification of Argonautes in nematodes: an
expanding universe. Biochemical Society Transactions 41: 881-86.
Capella-Gutiérrez S, Silla-Martínez JM, and Gabaldón T. 2009. trimAl: a tool for automated alignment
trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972-73.
Chang NC, Rovira Q, Wells J, Feschotte C, and Vaquerizas JM. 2022. Zebrafish transposable elements
show extensive diversification in age, genomic distribution, and developmental expression. Genome
Res 32: 1408-23.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
24
Charlesworth B and Langley CH. 1989. The population genetics of Drosophila transposable elements.
Annu Rev Genet 23: 251-87.
Chen Z, et al. 2019. De novo assembly of the goldfish (Carassius auratus) genome and the evolution of
genes after whole-genome duplication. Sci Adv 5: eaav0547.
Chernomor Olga, von Haeseler Arndt, and Minh Bui Quang. 2016. Terrace Aware Data Structure for
Phylogenomic Inference from Supermatrices. Systematic Biology 65: 997-1008.
Chung HR, Löhr U, and Jäckle H. 2007. Lineage-specific expansion of the zinc finger associated domain
ZAD. Mol Biol Evol 24: 1934-43.
Cui Jun, Sartain Caroline V., Pleiss Jeffrey A., and Wolfner Mariana F. 2013. Cytoplasmic
polyadenylation is a major mRNA regulator during oogenesis and egg activation in Drosophila.
Developmental Biology 383: 121-31.
Czech B, Preall JB, McGinn J, and Hannon GJ. 2013. A transcriptome-wide RNAi screen in the
Drosophila ovary reveals factors of the germline piRNA pathway. Mol Cell 50: 749-61.
Czech B and Hannon GJ. 2016. One Loop to Rule Them All: The Ping-Pong Cycle and piRNA-Guided
Silencing. Trends Biochem Sci 41: 324-37.
Degnan BM, Vervoort M, Larroux C, and Richards GS. 2009. Early evolution of metazoan transcription
factors. Curr Opin Genet Dev 19: 591-99.
Dobin Alexander, Davis Carrie A., Schlesinger Felix, Drenkow Jorg, Zaleski Chris, Jha Sonali, Batut
Philippe, Chaisson Mark, and Gingeras Thomas R. 2013. STAR: ultrafast universal RNA-seq
aligner. Bioinformatics 29: 15-21.
Ecco Gabriela, Imbeault Michael, and Trono Didier. 2017. KRAB zinc finger proteins. Development 144:
2719-29.
Eddy Sean R. 2011. Accelerated Profile HMM Searches. PLoS Computational Biology 7: e1002195.
Emerson Ryan O. and Thomas James H. 2009. Adaptive evolution in zinc finger transcription factors.
PLoS Genetics 5:
Fabry MH, Falconio FA, Joud F, Lythgoe EK, Czech B, and Hannon GJ. 2021. Maternally inherited
piRNAs direct transient heterochromatin formation at active transposons during early Drosophila
embryogenesis. Elife 10: e68573.
Fedotova AA, Bonchuk AN, Mogila VA, and Georgiev PG. 2017. C2H2 zinc finger proteins: the largest
but poorly explored family of higher eukaryotic transcription factors. Acta Naturae (англоязычная
версия) 9: 1965-84.
Felsenstein Joseph. 1985. Phylogenies and the comparative method. The American Naturalist 125: 1-15.
Fernandes Jason D, et al. 2018. KRAB Zinc Finger Proteins coordinate across evolutionary time scales to
battle retroelements.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
25
Flynn Jullien M., Hubley Robert, Goubert Clément, Rosen Jeb, Clark Andrew G., Feschotte Cédric, and
Smit Arian F. 2019. RepeatModeler2: automated genomic discovery of transposable element
families.
Font J and Mackay JP. 2010. Beyond DNA: zinc finger domains as RNA-binding modules. Methods Mol
Biol 649: 479-91.
Fricke Ron, Eschmeyer William, and Fong Jon David. 2020. Eschmeyer’s Catalog of Fishes.
Fueyo R, Judd J, Feschotte C, and Wysocka J. 2022. Roles of transposable elements in the regulation of
mammalian transcription. Nat Rev Mol Cell Biol 23: 481-97.
Gamez S, Srivastav S, Akbari OS, and Lau NC. 2020. Diverse Defenses: A Perspective Comparing
Dipteran Piwi-piRNA Pathways. Cells 9: E2180.
Goodstadt L, Heger A, Webber C, and Ponting CP. 2007. An analysis of the gene complement of a
marsupial, Monodelphis domestica: evolution of lineage-specific genes and giant chromosomes.
Genome Res 17: 969-81.
Goubert C, Modolo L, Vieira C, ValienteMoro C, Mavingui P, and Boulesteix M. 2015. De novo
assembly and annotation of the Asian tiger mosquito (Aedes albopictus) repeatome with dnaPipeTE
from raw genomic reads and comparative analysis with the yellow fever mosquito (Aedes aegypti).
Genome Biol Evol 7: 1192-205.
Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference
genome. Nat Biotechnol 29: 644-52.
Grewal SI. 2010. RNAi-dependent formation of heterochromatin and its diverse functions. Curr Opin
Genet Dev 20: 134-41.
Guo Yixuan, Gert Krista R, Lebedeva Svetlana, Potok Magdalena E, Wike Candice L, Grow Edward J,
Ketting René F, Pauli Andrea, and Cairns Bradley R. 2021. Maternally-inherited anti-sense piRNAs
antagonize transposon expression in teleost embryos.
Hadzhiev Y, et al. 2019. A cell cycle-coordinated Polymerase II transcription compartment encompasses
gene expression before global genome activation. Nat Commun 10: 691.
Hadzhiev Yavor, et al. 2021. The miR-430 locus with extreme promoter density is a transcription body
organizer, which facilitates long range regulation in zygotic genome activation.
Haig David. 2016. Transposable elements: Self-seekers of the germline, team-players of the soma.
BioEssays 38: 1158-66.
Hall Ira M., Shankaranarayana Gurumurthy D., Noma Ken-ichi, Ayoub Nabieh, Cohen Amikam, and
Grewal Shiv I. S. 2002. Establishment and Maintenance of a Heterochromatin Domain. Science
297: 2232-37.
Haring NL, van Bree EJ, Jordaan WS, Roels JRE, Sotomayor GC, Hey TM, White FTG, Galland MD,
Smidt MP, and Jacobs FMJ. 2021. ZNF91 deletion in human embryonic stem cells leads to ectopic
activation of SVA retrotransposons and up-regulation of KRAB zinc finger gene clusters. Genome
Res
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
26
Heger P, Zheng W, Rottmann A, Panfilio KA, and Wiehe T. 2020. The genetic factors of bilaterian
evolution. Elife 9: e45530.
Hoang Diep Thi, Chernomor Olga, von Haeseler Arndt, Minh Bui Quang, and Vinh Le Sy. 2018.
UFBoot2: Improving the Ultrafast Bootstrap Approximation. Molecular Biology and Evolution 35:
518-22.
Hoff Katharina J. and Stanke Mario. 2018. Predicting Genes in Single Genomes with AUGUSTUS.
Current Protocols in Bioinformatics e57.
Howe K, Schiffer PH, Zielinski J, Wiehe T, Laird GK, Marioni JC, Soylemez O, Kondrashov F, and
Leptin M. 2016. Structure and evolutionary history of a large family of NLR proteins in the
zebrafish. Open Biol 6: 160009.
Howe Kerstin, et al. 2013. The zebrafish reference genome sequence and its relationship to the human
genome. Nature 496: 498-503.
Huang W, Li L, Myers JR, and Marth GT. 2012. ART: a next-generation sequencing read simulator.
Bioinformatics 28: 593-94.
Hughes Lily C., et al. 2018. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on
transcriptomic and genomic data. Proceedings of the National Academy of Sciences 115: 6249-54.
Huntley S, Baggott DM, Hamilton AT, Tran-Gyamfi M, Yang S, Kim J, Gordon L, Branscomb E, and
Stubbs L. 2006. A comprehensive catalog of human KRAB-associated zinc finger genes: insights
into the evolutionary history of a large family of transcriptional repressors. Genome Res 16: 669-77.
Imbeault Michaël, Helleboid Pierre-Yves, and Trono Didier. 2017. KRAB zinc-finger proteins contribute
to the evolution of gene regulatory networks. Nature 543: 550-54.
Jacobs Frank M. J., Greenberg David, Nguyen Ngan, Haeussler Maximilian, Ewing Adam D., Katzman
Sol, Paten Benedict, Salama Sofie R., and Haussler David. 2014. An evolutionary arms race
between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 516: 242-45.
Jumper J, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596: 583-89.
Jurka J, Kohany O, Pavlicek A, Kapitonov VV, and Jurka MV. 2004. Duplication, coclustering, and
selection of human Alu retrotransposons. Proc Natl Acad Sci U S A 101: 1268-72.
Kaplan T, Friedman N, and Margalit H. 2005. Ab initio prediction of transcription factor targets using
structural knowledge. PLoS Comput Biol 1: e1.
Kasinathan Bhavatharini, Colmenares Serafin U, McConnell Hannah, Young Janet M, Karpen Gary H,
and Malik Harmit S. 2020. Innovation of heterochromatin functions drives rapid evolution of
essential ZAD-ZNF genes in Drosophila. eLife 9:
Katoh K. and Standley D. M. 2013. MAFFT Multiple Sequence Alignment Software Version 7:
Improvements in Performance and Usability. Molecular Biology and Evolution 30: 772-80.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
27
Keller O, Kollmar M, Stanke M, and Waack S. 2011. A novel hybrid gene prediction method employing
protein multiple sequence alignments. Bioinformatics 27: 757-63.
Kent Tyler V., Uzunović Jasmina, and Wright Stephen I. 2017. Coevolution between transposable
elements and recombination. Philosophical Transactions of the Royal Society B: Biological
Sciences 372: 20160458.
Klug A. 2010. The discovery of zinc fingers and their development for practical applications in gene
regulation and genome manipulation. Q Rev Biophys 43: 1-21.
Kosiol C, Holmes I, and Goldman N. 2007. An empirical codon model for protein sequence evolution.
Mol Biol Evol 24: 1464-79.
Kumar S, Suleski M, Craig JM, Kasprowicz AE, Sanderford M, Li M, Stecher G, and Hedges SB. 2022.
TimeTree 5: An Expanded Resource for Species Divergence Times. Mol Biol Evol msac174.
Laue Kathrin, Rajshekar Srivarsha, Courtney Abigail J, Lewis Zachary A, and Goll Mary G. 2019. The
maternal to zygotic transition regulates genome-wide heterochromatin establishment in the
zebrafish embryo. Nature Communications 10:
Li HD, Lin CX, and Zheng J. 2022. GTFtools: a software package for analyzing various features of gene
models. Bioinformatics 38: 4806-8.
Liu Hui, Chang Li-Hsin, Sun Younguk, Lu Xiaochen, and Stubbs Lisa. 2014. Deep Vertebrate Roots for
Mammalian Zinc Finger Transcription Factor Subfamilies. Genome Biology and Evolution 6: 510-
25.
Love Michael I, Huber Wolfgang, and Anders Simon. 2014. Moderated estimation of fold change and
dispersion for RNA-seq data with DESeq2. Genome biology 15: 550.
Löytynoja A. 2014. Phylogeny-aware alignment with PRANK. Methods Mol Biol 1079: 155-70.
Ma Q, et al. 2021. A mosquito small RNA genomics resource reveals dynamic evolution and host
responses to viruses and transposons. Genome Res 31: 512-28.
Mallick S, Gnerre S, Muller P, and Reich D. 2009. The difficulty of avoiding false positives in genome
scans for natural selection. Genome Res 19: 922-33.
Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, and
Shinkai Y. 2010. Proviral silencing in embryonic stem cells requires the histone methyltransferase
ESET. Nature 464: 927-31.
McLaughlin RN and Malik HS. 2017. Genetic conflicts: the usual suspects and beyond. J Exp Biol 220:
6-17.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, and Lanfear R.
2020. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic
Era. Mol Biol Evol 37: 1530-34.
Mistry J, et al. 2021. Pfam: The protein families database in 2021. Nucleic Acids Res 49: D412-19.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
28
Molparia B, Goyal K, Sarkar A, Kumar S, and Sundar D. 2010. ZiF-Predict: a web tool for predicting
DNA-binding specificity in C2H2 zinc finger proteins. Genomics Proteomics Bioinformatics 8:
122-26.
Mora C, Tittensor DP, Adl S, Simpson AG, and Worm B. 2011. How many species are there on Earth and
in the ocean. PLoS Biol 9: e1001127.
Najafabadi Hamed S, et al. 2015. C2H2 zinc finger proteins greatly expand the human regulatory lexicon.
Nature Biotechnology 33: 555-62.
Najafabadi Hamed S., Garton Michael, Weirauch Matthew T., Mnaimneh Sanie, Yang Ally, Kim Philip
M., and Hughes Timothy R. 2017. Non-base-contacting residues enable kaleidoscopic evolution of
metazoan C2H2 zinc finger DNA binding. Genome Biology 18: 1-15.
Nowick K, Fields C, Gernat T, Caetano-Anolles D, Kholina N, and Stubbs L. 2011. Gain, loss and
divergence in primate zinc-finger genes: a rich resource for evolution of gene regulatory differences
between species. PLoS One 6: e21553.
Panfilio KA, et al. 2019. Molecular evolutionary trends and feeding ecology diversification in the
Hemiptera, anchored by the milkweed bug genome. Genome Biol 20: 64.
Panyushev N, Okorokova L, Danilov L, and Adonin L. 2021. Pattern of Repetitive Element Transcription
Segregate Cell Lineages during the Embryogenesis of Sea Urchin Strongylocentrotus purpuratus.
Biomedicines 9: 1736.
Parisot N, et al. 2021. The transposable element-rich genome of the cereal pest Sitophilus oryzae. BMC
Biol 19: 241.
Persikov AV and Singh M. 2014. De novo prediction of DNA-binding specificities for Cys2His2 zinc
finger proteins. Nucleic Acids Res 42: 97-108.
Platt RN, Vandewege MW, and Ray DA. 2018. Mammalian transposable elements and their impacts on
genome evolution. Chromosome Res 26: 25-43.
Pontis J, Pulver C, Playfoot CJ, Planet E, Grun D, Offner S, Duc J, Manfrin A, Lutolf MP, and Trono D.
2022. Primate-specific transposable elements shape transcriptional networks during human
development. Nat Commun 13: 7178.
Pontis Julien, Planet Evarist, Offner Sandra, Turelli Priscilla, Duc Julien, Coudray Alexandre, Theunissen
Thorold W., Jaenisch Rudolf, and Trono Didier. 2019. Hominoid-Specific Transposable Elements
and KZFPs Facilitate Human Embryonic Genome Activation and Control Transcription in Naive
Human ESCs. Cell Stem Cell 24: 724-735.e5.
Potireddy Santhi, Vassena Rita, Patel Bela G., and Latham Keith E. 2006. Analysis of polysomal mRNA
populations of mouse oocytes and zygotes: Dynamic changes in maternal mRNA utilization and
function. Developmental Biology 298: 155-66.
Quinlan Aaron R. and Hall Ira M. 2010. BEDTools: a flexible suite of utilities for comparing genomic
features. Bioinformatics 26: 841-42.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
29
Rice P, Longden I, and Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software
Suite. Trends Genet 16: 276-77.
Rodriguez-Terrones Diego and Torres-Padilla Maria-Elena. 2018. Nimble and Ready to Mingle:
Transposon Outbursts of Early Development. Trends in Genetics 34: 806-20.
Rowe HM, et al. 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463: 237-
40.
Sahney S, Benton MJ, and Ferry PA. 2010. Links between global taxonomic diversity, ecological
diversity and the expansion of vertebrates on land. Biol Lett 6: 544-47.
Sasaki Mariko, Lange Julian, and Keeney Scott. 2010. Genome destabilization by homologous
recombination in the germ line. Nature Reviews Molecular Cell Biology 11: 182-95.
Senft AD and Macfarlan TS. 2021. Transposable elements shape the evolution of mammalian
development. Nat Rev Genet
Seroussi Uri, Lugowski Andrew, Wadi Lina, Lao Robert X., Willis Alexandra R., Zhao Winnie, Sundby
Adam E., Charlesworth Amanda G., Reinke Aaron W., and Claycomb Julie M. 2022. A
Comprehensive Survey of <i>C. elegans</i> Argonaute Proteins Reveals Organism-wide Gene
Regulatory Networks and Functions. bioRxiv
Sienski G, Dönertas D, and Brennecke J. 2012. Transcriptional silencing of transposons by Piwi and
maelstrom and its impact on chromatin state and gene expression. Cell 151: 964-80.
Sniegowski Paul D. and Murphy Helen A. 2006. Evolvability. Current Biology 16: R831-34.
Tarlinton Rachael E., Meers Joanne, and Young Paul R. 2006. Retroviral invasion of the koala genome.
Nature 442: 79-81.
The PyMOL Molecular Graphics System. Version 2.0:
Thomas J. H. and Schneider S. 2011. Coevolution of retroelements and tandem zinc finger genes.
Genome Research 21: 1800-12.
To TH, Jung M, Lycett S, and Gascuel O. 2016. Fast Dating Using Least-Squares Criteria and
Algorithms. Syst Biol 65: 82-97.
Tollis M and Boissinot S. 2012. The evolutionary dynamics of transposable elements in eukaryote
genomes. Genome Dyn 7: 68-91.
Vacic V, Uversky VN, Dunker AK, and Lonardi S. 2007. Composition Profiler: a tool for discovery and
visualization of amino acid composition differences. BMC Bioinformatics 8: 211.
Varadi M, et al. 2022. AlphaFold Protein Structure Database: massively expanding the structural
coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50: D439-44.
Volpe Thomas A., Kidner Catherine, Hall Ira M., Teng Grace, Grewal Shiv I. S., and Martienssen Robert
A. 2002. Regulation of Heterochromatic Silencing and Histone H3 Lysine-9 Methylation by RNAi.
Science 297: 1833-37.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
30
Wei Kevin H-C, Chan Carolus, and Bachtrog Doris. 2021. Establishment of H3K9me3-dependent
heterochromatin during embryogenesis in Drosophila miranda. eLife 10:
White Richard J., et al. 2017. A high-resolution mRNA expression time course of embryonic
development in zebrafish. eLife 6:
Winata CL, et al. 2018. Cytoplasmic polyadenylation-mediated translational control of maternal mRNAs
directs maternal-to-zygotic transition. Development 145:
Wolf D and Goff SP. 2009. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458:
1201-4.
Wolf G, de Iaco A, Sun MA, Bruno M, Tinkham M, Hoang D, Mitra A, Ralls S, Trono D, and Macfarlan
TS. 2020. KRAB-zinc finger protein gene expansion in response to active retrotransposons in the
murine lineage. Elife 9: e56337.
Wolf Gernot, Yang Peng, Füchtbauer Annette C., Füchtbauer Ernst-Martin, Silva Andreia M., Park
Chungoo, Wu Warren, Nielsen Anders L., Pedersen Finn S., and Macfarlan Todd S. 2015. The
KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous
retroviruses. Genes & Development 29: 538-54.
Wolfe SA, Nekludova L, and Pabo CO. 2000. DNA recognition by Cys2His2 zinc finger proteins. Annu
Rev Biophys Biomol Struct 29: 183-212.
Xu P, et al. 2014. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat
Genet 46: 1212-19.
Yang Z. 2007. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Molecular Biology and
Evolution 24: 1586-91.
Yu Tianxiong, Koppetsch Birgit S., Pagliarani Sara, Johnston Stephen, Silverstein Noah J., Luban
Jeremy, Chappell Keith, Weng Zhiping, and Theurkauf William E. 2019. The piRNA Response to
Retroviral Invasion of the Koala Genome. Cell 179: 632-643.e12.
Zeller P, Padeken J, van Schendel R, Kalck V, Tijsterman M, and Gasser SM. 2016. Histone H3K9
methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA
hybrid-associated repeat instability. Nat Genet 48: 1385-95.
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 30, 2022. ; https://doi.org/10.1101/2022.11.29.518450doi: bioRxiv preprint
