
Emory University IRB
STANDARD OPERATING
PROCEDURES

Table of Contents
Page 2 of 273
Table of Contents
Table of Contents ............................................................................................................................................................. 2
ADMINISTRATIVE ............................................................................................................................ 5
SOP Portfolio Modifications .............................................................................................................................................. 5
Listserv Responsibilities .................................................................................................................................................... 9
Mass Email Listerv Management .................................................................................................................................... 13
IRB Staff Study Checklists/Worksheets/Other documents Updates ............................................................................... 18
Onboarding New IRB Staff .............................................................................................................................................. 19
Evaluating IRB Staff Performance ................................................................................................................................... 21
Inquiries & Complaints received about IRB submissions ................................................................................................ 25
IRB Staff Meeting Facilitation .......................................................................................................................................... 28
COLLABORATIVE RESEARCH / CENTRAL IRBS ............................................................................... 29
External Collaborators Included in Initial Submissions or in Modifications to Approved Non-Exempt Research ........... 29
Obtaining Access to WIRB and NCI CIRB ......................................................................................................................... 32
WCG Listserv Duties ........................................................................................................................................................ 33
NCI CIRB studies processing ............................................................................................................................................ 35
Reportable New Information (RNI) Submission Notification Process for External IRB Studies ....................................... 42
Vetting NIH Single IRB Plans ............................................................................................................................................ 46
XIRB Study Processing When Emory Relying on An External IRB Other than the NCI CIRB ............................................ 48
XIRB Study Processing for Studies Transitioning from Emory IRB Approval to External IRB Review .............................. 54
Closing Out Multi-Site Studies with Central or Single IRB Review ................................................................................... 55
Issuing Partial HIPAA Waivers for XIRB Studies ............................................................................................................... 57
Processing New Studies When Emory is the Single IRB of Record (sIRB) for a Multi-Site Study ..................................... 58
Processing Modifications When Emory is the Single IRB of Record (sIRB) for a Multi-Site Study ................................... 62
Processing CRs When Emory is the Single IRB of Record (sIRB) for a Multi-Site Study ................................................... 63
IRB MEMBER MANAGEMENT ....................................................................................................... 64
IRB Member Onboarding ................................................................................................................................................ 65
QA AND EDUCATION ..................................................................................................................... 69
Acknowledgments & Noncompliance Determinations Made by Senior Team Q Staff ................................................... 69
IRB Noncompliance ......................................................................................................................................................... 71
IRB Team Q CAPA Follow Up ........................................................................................................................................... 75
CMTE Q Meeting Facilitation .......................................................................................................................................... 77
Communication of Report of Internal Study Subject Death ............................................................................................ 80
Review of Single Use, Expanded Access of Unapproved Drugs or Devices ..................................................................... 82
External Webinars presented by Team Q ....................................................................................................................... 87

Table of Contents
Page 3 of 273
Process of Review of Consent Process Errors after Notification from OCR .................................................................... 89
Informed Consent Monitoring SOP ................................................................................................................................. 92
Internal QA/QI review of documents after IRB Review .................................................................................................. 94
Letters after FB with PIs, OHRP and FDA after SNC, CNC and UP determinations .......................................................... 96
Reportable new information submission Review Process .............................................................................................. 97
Education and Quality Assurance Team Mission and Process ...................................................................................... 105
IRB Record Review of Studies approved by the Emory IRB ........................................................................................... 107
Review of Safety Reports submitted by sponsors holding an IDE ................................................................................. 110
Routing External UPs (FDA Regulated) .......................................................................................................................... 112
S-I SUBMISSION MANAGEMENT ............................................................................................... 115
New study screen process for S-I studies ...................................................................................................................... 115
CR screen process for S-I studies .................................................................................................................................. 118
Modification (MOD) submission screen process for S-I studies .................................................................................... 120
Closeout submission screen process for S-I studies ...................................................................................................... 123
S-I, Single IRB Study Screening Process ......................................................................................................................... 125
STUDY MANAGEMENT ................................................................................................................ 127
Not Research/ Not-Human-Subjects/Not-Human-Subjects-Research/Not-Engaged .................................................... 127
MODIFICATIONS .......................................................................................................................... 132
Adding/Removing Study Personnel .............................................................................................................................. 132
Modifications- Processing from Preliminary Analysis through Approval ...................................................................... 140
Modifications: Mods Indicating Increased Risk ............................................................................................................. 146
NEW STUDIES .............................................................................................................................. 148
Pre-Review options and Ancillary Review Information ................................................................................................. 148
eIRB Processing of New Study Applications- Preliminary Analysis through Approval................................................... 164
Naming Conventions for eIRB Studies........................................................................................................................... 171
RDRC Studies ................................................................................................................................................................. 172
Translation of Informed Consent Documents ............................................................................................................... 174
Training Verification ...................................................................................................................................................... 177
Electronic documentation of informed consent via “electronic signature” or “digital signature”) .............................. 180
Mobile Devices and Mobile Medical Apps Used in Research ........................................................................................ 187
Certificate of Confidentiality Process for non-federally funded studies ....................................................................... 193
Data sharing certifications including genomic data sharing .......................................................................................... 199
COI: Handling Studies with Study Team Conflict of Interest ......................................................................................... 201
Institutional Conflict of Interest .................................................................................................................................... 207
Cost Option for Clinical Trial Agreements and ICFs ....................................................................................................... 210

Table of Contents
Page 4 of 273
Sensitive Study Status ................................................................................................................................................... 212
Imaging Studies ............................................................................................................................................................. 214
How to Handle GWAS Data Use Certification Requests ................................................................................................ 216
Humanitarian Device Exemption (HDE) Studies ............................................................................................................ 217
ResearchMatch.org as a recruitment tool .................................................................................................................... 221
Checking Biosafety Approval Status .............................................................................................................................. 222
Processing Studies that will use Deception or Incomplete Disclosure .......................................................................... 223
St. Joseph and John’s Creek study site process ............................................................................................................. 226
REMS study review ....................................................................................................................................................... 229
Prisoner Studies: Handling of New/Modification/Renewal Submissions when Prisoners are Subjects (Application of
Subpart C) ..................................................................................................................................................................... 231
VA Studies with non-VA Sites – IRB Submission Requirements .................................................................................... 236
Determinations and Reviews by IRB Staff ..................................................................................................................... 238
Categories of Research Reviewable by IRB Staff as IRB Designated Members ............................................................. 240
DURING STUDY CONDUCT .......................................................................................................... 247
Over-Enrollment Via Consent (No Research Activities including during Screening) ..................................................... 247
Transferring Study Participants Between Study Sites ................................................................................................... 249
Fixing Errors in eIRB System .......................................................................................................................................... 251
Continuing Review Processing-Preliminary Analysis through Approval ........................................................................ 254
Continuing Review: Applying 30-day window ............................................................................................................... 260
Continuing Review: REs/PDs/Noncompliance and Monitor Reports ............................................................................ 262
Continuing Review: Processing study staff noncompliance with CITI and Clinical Research Training (formerly Key
Concepts/Intro to CR) requirements ............................................................................................................................. 265
CLOSE OUT .................................................................................................................................. 267
Close-Out Process ......................................................................................................................................................... 267
Informing Teams of Study Closure ................................................................................................................................ 272

Table of Contents
Page 5 of 273
ADMINISTRATIVE
SOP Title:
SOP Portfolio Modifications
SOP Category:
Administrative
Established:
8/29/2013
Last Revision:
4/22/2022
PURPOSE
The purpose of this document is to describe the process of adding or modifying approved SOPs,
Guidance, and Policies (‘IRB documents’) into the designated H drive area.
SCOPE OF SOP
The SOP applies to the SOPs, Guidance, and Policies affecting the Emory IRB.
RESPONSIBILITIES
• IRB Designated SOP Manager: Add approved IRB documents to the designated H drive area.
• IRB Director: Gives final approval of any new SOP in the designated H drive area.
• IRB SOP Sub-committee: Identifies the need for the creation or modification of existing IRB
documents.
• Huron: periodically, the eIRB system vendor (Huron) will send updates to improve the electronic
system. These updates may include updates based on changes in the guidance or policies affecting
human subjects' research. These changes will come in the form of updates to the electronic system
and the Huron Toolkit.
PROCEDURE
Note: The official SOP Portfolio is a pdf document that is uploaded to our website documents
folder. There’s no link to it from our website itself to keep the document accessible only to the IRB staff.
The direct link to the portfolio is: http://www.irb.emory.edu/documents/SOP%20Portfolio.pdf
After Huron updates
1. Periodically, Huron will update the electronic system and Toolkit. The Toolkit is the group of
regulatory documents Huron created to be used with the electronic system. At Emory, we are only
implementing a selection of the checklists and worksheets from the Toolkit.
2. When a Huron update affects our SOPs, P&Ps, checklist, and worksheets, we will update these
documents in the next 30 business days, if required.
For SOP portfolio suggestions (staff)
1. To suggest changes to an SOP, copy the SOP from the SOP portfolio document in word.
2. Track changes and alert the IRB Designated SOP Manager. Save copy under
H:\General\Admin IRB Documents\SOP Portfolio\SOPs in process_Pre Approval_Not Ready to Add in
to the Portfolio Yet\In progress
3. Ask a member of the staff leadership team to review the change. For a new SOP, the IRB Director
should approve it before adding it to the SOP portfolio.
4. After the Director or staff leadership member, as applicable, approve the new or changes to an
existing SOP, move the document to the folder entitled H:\General\Admin IRB Documents\SOP

Table of Contents
Page 6 of 273
Portfolio\SOP Portfolio Source Files & Where TLs track in ready to go live changes\Revised SOPs
Already Approved and Ready to track in to the SOP and go live
Instructions for the SOP Portfolio manager
1. Track and make the approved changes to the Word document portfolio (H:\General\Admin IRB
Documents\SOP Portfolio\SOP Portfolio Source Files\SOP Portfolio.docx)
a. Some changes could have been made to the clean copy, ‘clean copy for future tracked
changes’ of the current portfolio located at H:\General\Admin IRB Documents\SOP
Portfolio\SOP Portfolio Source Files
b. For other changes located under the “Ready for Add to Portfolio” folder, copy and paste just
the body text of the new/revised SOP into the main portfolio; copying the header and the log
of changes often led to formatting issues.
c. Add a new version date on the SOP that should be the date of the release of the SOP to the
staff.
d. Under “Log of Significant changes” add the date of the portfolio revisions (same as the date on
c) and describe the changes. Be as descriptive but succinct as possible.
e. After all the changes are made, remember to update the table of contents so that the page
numbers are accurate. This is done automatically by simply right-clicking on the table of
contents, click on “update field, and then on “Update entire table”
f. Review the table of contents and delete any subheaders.
g. Keep a copy of the tracked version, and create a new, clean version. PDF the clean version.
2. Update the online SOP portfolio with the revised, clean PDF version:
a. Log in to Cascade: https://cascade.emory.edu
b. Select “RE Institutional Review Board – IRB” from the dropdown menu at the very top of the
first window
c. From the left-hand menu, navigate to Base Folder/documents/SOP Portfolio.pdf
d. Go to the “Edit” tab
e. Select the revised PDF version of the portfolio, then click Submit

Table of Contents
Page 7 of 273
f. After replacing with the new version, click on “Publish”.
3. After about a minute or so, check the online SOP link to make sure that the most recent version was
successfully uploaded. You may need to press your browser’s refresh button to clear the cache
(force it to “forget” the old version)
4. Email or note in teams channel chat the IRB staff, letting them know about the changes, with a copy
of the tracked SOP portfolio. Direct the Pod leaders to review these changes at their next meeting
and add them to the next IRB staff meeting for in-depth review if needed.
See below an example of such an email/teams chat notification:
Subject: Changes to SOP Portfolio: November 1, 2018
Hi everyone,
Please, review the latest changes for the SOP portfolio, to keep up-to-date with new processes, as
applicable.
Remember to refresh your browser in case you do not see the changes.
Sr. RPAs: PLEASE SAVE THIS IN THE IMPORTANT NEWS TAB OF YOUR POD REPORT TO REVIEW DURING THE NEXT POD
MEETING. FEEL FREE TO REVIEW ONLY THE SOPS AFFECTING YOUR TEAM.
See the attached document for additional details on these changes. The following SOPs were modified,
added, or deleted:
Changes to SOPs (see changes in attached tracked changes document)
• SOP Portfolio Modifications- Updating to reflect current practice
• Meeting Facilitation Responsibilities- Added that Meeting Materials be added as supporting
documents to the Submit RNI Committee Review activity for RNIs.
• Modifications- Processing from Preliminary Analysis through Approval- Changes in the process to
review contingency reviews.
• eIRB Processing of New Study Applications- Preliminary Analysis through Approval- Changes in the
process to review contingency reviews.

Table of Contents
Page 8 of 273
• Certificate of Confidentiality Process in non-federal studies- Updated to follow new Online
Certificate of Confidentiality System User Guide dated 06/25/2020.
• Continuing Review Processing-Preliminary Analysis through Approval- Changes in the contingency
review process
New SOP
• Advarra Study Processing from Submission to Approval
Let me know if you have any questions,
NAME
5. Save a copy of the email or teams chat notification sent to the staff under H:\General\Admin IRB
Documents\SOP Portfolio\Emails or Notifications sent to staff about portfolio changes
6. Save the tracked and PDF versions of the SOP in the archived portfolios folder located at
H:\General\Admin IRB Documents\SOP Portfolio\Archived Portfolios
PROCESS FLOW
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/5/2021
Updating to reflect current practice
5/24/2021
Added more details in this process, updating graphics
1/21/2022
Removed section about SOP subcommittee & archived log of changes over a year old & updated
folder location for archived portfolios
4/22/2022
Updated based on current practice
IRB document
is created or
modified
Associate or Assistant Director
reviews document for accuracy
with current IRB or Emory
policies, or federal regulations
Reviewed version is
sent to IRB director
for final review and
approval
Final version is sent
to IRB designated
person to be added
to H Drive

Table of Contents
Page 9 of 273
SOP Title: Listserv Responsibilities
SOP Category:
Administrative
Established:
9/4/2013
Last Revision:
08/15/2024
NOTE: Listserv responses MUST be given the same day or at the latest the next day, with no exceptions.
If you can’t fully respond, at least email to say that you are working on it (or have passed it on to
someone, see “Required Procedures for Handling Listserv” below). Listserv day starts at 4 pm the day
before the assigned day. The shift ends at 3:59 pm on the assigned day. For example, for someone with
a Wednesday assigned day, the shift will start at 4 pm on Tuesday and end at 3:59 pm on Wednesday.
Types of Emails
How to respond/Action taken
Complaints from
participants
Forward to Team Q (Shara Karlebach); cc Director
Required Procedures for Handling
Listserv
If you send a response to a listserv
email
Not required but preferred practice: Bcc yourself on the
response and then save the blind copy to your “Listserv Duties”
folder to more easily keep track of what you have done and can
let other IRB staff know what has been done if they have
questions. Alternatively, save the sent email into your Listerv
duties folder.
If you forward the email to another
analyst
ALWAYS do so by clicking “Reply” and CC’ing the analyst. The
reply should let the sender know you’ve received their email
and are sending it to [analyst] to handle it. That way the sender
knows who’s responsible for their inquiry. If there is an
attached document, forward to assigned analyst in separate
email.
If you cannot cover listserv due to
illness or vacation
Communicate with another listserv staff member to ensure
coverage (e.g., switch days for that week) and inform one of the
Directors (or direct supervisor)
CITI Completion Reports
Just move to your “CITI Completion Reports” folder under inbox
(best to set up a “Rule” so Outlook does this automatically)
If you handle listserv on a
University Holiday
Complete tasks from your assigned day when you return to the
office.
If designated person [see contact
grid] from Emory Admin Assistant
is unavailable during your listserv
day
Complete Emory Admin Assistant Designated Person’s tasks.

Table of Contents
Page 10 of 273
Compliance or
safety event
related
Send it to Team Q (Shara Karlebach, , Jackson Parker, Briana Rotterman). If a
specific study is mentioned, CC the owner of the study.
Contact IRB Staff
Activity
Emails with this Subject line are generated when someone logs a comment in a
study before the study is assigned to an analyst.
If the comment is informational and not at all time-sensitive – someone will see
it once it has been assigned – you don’t need to do anything about this type of
email – Bcc yourself to indicate that no response was made and save to your
“Listserv duties” folder.
If the comment is a question or request for assistance that requires a response,
you will need to 1) either send an email as a response, 2) refer the issue to
someone else, or 3) respond via logged comment. Remember to Bcc yourself to
keep track of what was done.
Corp_IRB_Options.
xls from ORA-IT or
OSP
The subject line is “Corp_IRB_Options – Records Found.” Note: Proposal ID is
NOT the IRB file number. Listserv person is not expected to do anything with
this email.
PRMC approvals
(*)
Forward to the designated person (see contact grid). The designated person will
upload as a comment in eIRB. In addition, the designated person will upload
DSMB plans, when submitted via listserv.
The comment should include whether a study was approved, pended, or
disapproved. Distinguish between DSMB plans and PRMC approval.
DSMC Plan
approvals
Disregard any that are received.
eIRB account
requests
Direct to the IRB Website: https://irb.emory.edu/guidance/faqs/eirb-saas.html
Emails containing
identifiable health
information, like
the name of the
study subject.
Delete the email from all folders (including the "Trash") and send an email to all
IRB staff (do NOT include the PHI) OR post on IRB-Staff | General | Microsoft
Teams and request that they do the same (identify the email by sender or
subject).
Fee/Cost
Questions
Refer them to OCR’s memo on research fees located here:
https://ocr.emory.edu/secure/emory_university_standard_research_study_fee
s_signed-memo_7_18_2022_final-1.pdf
(pull-down ‘Research fees’). Otherwise, Sheila O’Neal, the finance supervisor
for OCR, has offered to answer any questions about IRB fees or fee schedules.
FWA, IRB
registration
questions
First check the IRB website for the answer (or direct the
questioner):https://irb.emory.edu/about/index.html . If still unclear, forward
to IRB Director.
IRB authorization
agreements,
collaborating with
other institutions
Forward to reliance team at irb.reliance@emory.edu

Table of Contents
Page 11 of 273
Membership List
Requests/Roster
Requests
The IRB does not provide rosters generally. Sponsors may use the compliance
letter to confirm that no conflicted members take part in IRB reviews. Refer to
Contact Us section on the IRB website for a partial roster (IRB Members):
https://irb.emory.edu/about/contact/irb-members.html
If a pdf is requested, they can copy and paste the information from the website
into Word.
Need a copy of CITI
completion report
Give them the web address of the CITI website. If they then say they still can’t
access their report, we can send it to them (Contact ADs or Team Q for
assistance). You can also assure them that our analysts check completions when
we screen studies, and we don’t need the certificate. You can also request a
reset on the CITI website for the member to get a temporary password sent to
their email.
Emails stating
Costs option
chosen for a
particular study (*)
Forward to the designated person (see contact grid). The designated person will
copy the attachment to their personal computer. The designated person should
open the corresponding study in eIRB and log a comment like “CTA Cost Option
is #___ per attached” and attach the file.
CHOA In Case of
Injury (ICOI) and
Cost Options
Forward to the designated person (see contact grid). The designated person will
copy the attachment to their personal computer. The designated person should
open the corresponding study in eIRB and log a comment like “ICOI Option is
#___ per attached” and attach the file.
“O-day Closeout
Notice for Your
Award”
You can ignore these.
Research Match
emails
Forward to IRB Director
Removal from or
addition to IRB
listserv ("blast")
requests
Forward to Briana Rotterman (Team Q)
Request for an in-
person
consultation
Take the first one, and then email office about additional requests during the
day. If the request is regarding reliance, send to the reliance Assistant Director.
Study-related
questions
Forward to the analyst who owns the submission. If the analyst isn’t available
on the day you are monitoring listserv, forward to Associate or Assistant
Director.
WCG IRB
questions,
notifications, and
forms
Do nothing if they come directly from WCG IRB. If they are emails from a study
team, please forward it to the analyst assigned to the study.
Other
If unsure how to handle requests that are not noted in this chart, forward to
Staff leadership and request assistance. Depending on the situation, you may
need to reply to the sender to acknowledge receipt and let them know that it is
being considered.

Table of Contents
Page 12 of 273
Emails from OIT
about security
reviews
An email will contain a report that should be uploaded under the study history.
If there are issues with finding an IRB number, please contact Shara Karlebach
for help. If the report indicates any critical issues, indicate that in the comment
as the study cannot be approved if still planning to use the software or app.
Voicemails from
IRB general
number received
via email
The person covering the Emory listserv is in charge of calling/emailing people
back. The listserv may forward the message to another person as done for
emails received in the listserv inbox, including inquiries about studies.
Sponsor not in the
list in eIRB
Review the Compass Sponsor list. If the requested sponsor is not on the
Compass Sponsor list, fill this form to request the sponsor to be added to the
system. Note that you will need the following information about the sponsor
organization to complete the form: legal name, address, phone number, and
URL. Let the study team know that after ORA IT receives this request, they will
need confirmation from OSP. If the requested sponsor is on the Compass
Sponsor List, fill out the form - can make hyperlink or use the same hyperlink in
the form. "In #1 enter 'yes,' then enter the customer number, which is in the
first column of the Compass Sponsor list next to the sponsor in question."
Reliance/Single IRB
Questions
Provide them a link to the Collaborative Research page on our website and copy
Not Research/
Not-Human-
Subjects/Not-
Human-Subjects-
Research/Not-
Engaged
See the SOP, Not Research/ Not-Human-Subjects/Not-Human-Subjects-
Research/Not-Engaged
(*) If the designated person is out of the office, the listserver is responsible for those tasks.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
9/11/2023
Replacing Enid Sullivan with link to contact grid. Archiving logs older then on year of significant
changes & clarification on what ‘analyst isn’t available’ means
06/12/2024
Updated to remove student workers, requests for powerchart access and added award closeout
notices.
08/15/2024
Removed reference to Jessica Blackburn as Team Q member. Updated all broken hyperlinks to IRB
and OCR websites.

Table of Contents
Page 13 of 273
SOP Title: Mass Email Listerv Management
SOP Category:
Administrative
Established:
11/6/2015
Last Revision:
10/14/2021
PURPOSE
Provides steps generate a list of recipients for a mass email from the IRB, formatting of the email, and
management of the listserv online system.
SCOPE
Applies only to IRBResearch-L listserv, not for IRB-L listserv (associated with irb@emory.edu
).
DEFINITIONS
• Blast: the email sent to all subscribers to the IRBResearch-L listserv.
PROCEDURES
Logging in for the first time
1. Go to http://listserv.cc.emory.edu/cgi-bin/wa?INDEX
2. If this is your first time – click on the blue text: get a new LISTSERV password.

Table of Contents
Page 14 of 273
Creating Email List
1. Run the custom ORAIT_All emails for mass mailingand export results
a. This pulls the emails of all eIRB accounts
b. eIRB does not remove old/expired accounts; there will be redundancies, but they are
irrelevant to the Blast process
2. Remove all columns except Emails
3. Save as Text (Tab delimited) to the E-mail blasts folder on H
a. General/QA Working Files/E-mail blasts/YEAR/MONTH
4. Go to the Emory Email List Service website
a. http://listserv.cc.emory.edu/
b. You should use SSO log in
5. From the top-left drop-down menu, select Subscriber Management

Table of Contents
Page 15 of 273
6. Select the Bulk Operations tab
7. Select the Add/Do Not Remove option and choose the exported data as the Input File
8. Import the file, checking for any error messages
9. Open the Remove from listserv spreadsheet in the Email Blasts folder
10. Remove all columns except emails and save as Text (Tab delimited) in /YEAR/MONTH folder
11. Return to the listserv website and select the Remove/Do Not Add option and import the new
Remove spreadsheet
12. Open the Add to listserv spreadsheet in the Email Blasts folder
13. Remove all columns except emails and save as Text (Tab delimited) in /YEAR/MONTH folder
14. Return to the listserv website and select the Add/Do Not Remove option and import the new
Add spreadsheet
Writing the Blast
1. Solicit topics from IRB staff
2. Use a previous blast or the blast template as a guide
3. Draft the blast in a Word document so that revisions can be more easily tracked
a. Formatting Components (for reference):
i. Title: Emory IRB Update at Cambria 26, Date at Cambria 12
ii. Table of Contents: Linking to individual sections; Title at Cambria 13 and Items
at Calibri 11
iii. Content: Titles linking back to TOC; Titles at Cambria 13, Body Text at Calibri 11
iv. Contact us: Includes IRB email, phone number, website, and physical office; Title
at Cambria 16, Body Text at Calibri 11
v. Unsubscribe instructions: Send unsubscribe email to listserv or request to IRB
email; Calibri Italic 10
b. TOC title link to sections
i. Highlight section title
ii. Right-click and select Hyperlink…

Table of Contents
Page 16 of 273
iii. Select Place in this Document
iv. Select the appropriate Heading
4. Content headings link back to the TOC
a. As above
b. Select the Things to Know heading
c. Repeat for each heading
5. To create new headings
a. Go to the View Tab
b. Select Outline view
c. Add desired text
d. Set as Level 2
6. Once the blast draft is ready, send it as an attachment to all the TLs for their input and revisions.
Unsubscribing instructions
1. Users can unsubscribe via two methods:
a. Send an email to listserv@listserv.cc.emory.edu
and type UNSUBSCRIBE IRBRESEARCH-L
in the body of the email, the subject should be left blank
i. This automatically removes them from the list and generates an email to
and to the listserv manager(s)
b. Send a request to [email protected]. Please include the email address you wish to have
removed
2. Add emails of individuals unsubscribing from listserv (via any method) to the master Remove
from listserv spreadsheet
a. If individuals still have eIRB accounts, their emails will be included in the original export
file, even if they have unsubscribed. This is why you must continuously update this list,
so that the unsubscription is saved.
Sending the Blast
1. Paste the drafted blast into an email. Make sure the banner image is centered in the email.
2. Ensure HTML is enabled
3. Double check that all links work correctly.
4. Send the email to irbresearch-l@listserv.emory.edu
Adding/Removing List Owners
1. Log in to the Emory Email List Service
2. From the List Management drop-down, select List Configuration, List Configuration Wizard.
then the List Maintenance tab
3. Add/Remove the relevant email from an Owner line
a. Be sure to use the netid@emory.edu
email address
4. Save
Archiving the Blast* on the IRB Website

Table of Contents
Page 17 of 273
*Only for news-related blasts. Webinar or other education-related blasts don’t need to be
archived.
1. Log in to Cascade
2. Navigate to the Education, Past News and Email Blasts, Past Email Blasts page
3. Copy and paste the information in a new section of the page. You will have to adjust formatting
a lot. You should remove all links except for links within blast news items to helpful documents.
4. You don’t need to include the Helpful Links or Unsubscribe information.
5. Title the new section according to the blast title.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
7/2/21
Updated report name for SaaS and log in instructions for SSO
10/14/21
Revised direction of drop down menu reference & archived log of updates over 1
year old

Table of Contents
Page 18 of 273
SOP Title:
IRB Staff Study Checklists/Worksheets/Other documents Updates
SOP Category:
Administrative
Established:
5/24/2021
Last Revision:
5/24/2021
PURPOSE
This SOP details the process of updating information in the IRB Staff study checklist and worksheets.
RESPONSIBILITIES
• IRB Director/Team Lead: makes changes to the checklists, worksheets or other documents and
informs the IRB Staff.
PROCEDURES
• These changes can only be made by team leads. The documents are stored here
(IRB-
Leadership/Documents/Linked docs-do not move or touch).
• To make changes to the documents, click on the ellipsis.
• Click on “open” and then “open in app”
• Save a local copy before making any changes.
• Click on “Review” and then “Track Changes”. Save a clean and tracked version of the document.
Make sure you update the document version in the footer. The clean version should have the same
name as the one in the one drive folder.
• Drag and drop the new, clean version of the folder in the one drive folder to replace the document.
Make sure the name of the document is the same as the one saved in the folder, so the document is
replaced.
• Announce the changes in Teams, under “IRB-Staff-To Remember”. Attached the tracked version of
the document.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 19 of 273
SOP Title:
Onboarding New IRB Staff
SOP Category:
Administrative
Established:
5/13/2015
Last Revision:
08/15/2024
PURPOSE
The purpose of this SOP is to document steps in the training of new employees. Resources for new hire
training can be found in the Education folder located here on the H drive H:\General\Education\Staff
Education New and Cont\New Staff Training and here http://www.irb.emory.edu/staff_training/
.
SCOPE
This SOP applies to training for all new IRB staff.
PROCEDURE
IRB leadership will announce the name of the new employee and their start date prior to the new
employee’s start date.
Pre-Hire Preparations
• Before the start date of the new employee, the supervisor will confirm that ORA has arranged for
computer equipment and monitors for the new employee and will obtain the new employee’s email
address.
• The supervisor will prepare the Go-To-Staff Training Sign-up Sheet and ask those who are assisting
with training (leadership and Sr. RPAs) to sign-up for the specific days to complete their training.
New Employee Training Program
• The supervisor will send the new employee the welcome email template with important links and
attachments including the training manual.
• The supervisor will meet with the new employee in person or on zoom and review the plan for the
first week and for training.
• The supervisor will schedule time to take the new hire to lunch and will schedule a virtual meet and
greet with other staff.
• Each day that training is scheduled, the trainer will send the zoom link for training and let the new
employee know what materials should be reviewed prior to training. The trainer will find relevant
IRB submissions to assign to the new employee for practice and will schedule time for follow up
questions and review the new employee’s work on the submissions.
• Sr. RPAs will schedule time to allow the new employee to shadow them performing reviews of
submissions and will be points of contact for questions as will the Director and ADs.
• Once the new employee has demonstrated competence reviewing continuing reviews,
modifications and new studies, a portfolio of studies will be assigned to the new employee.
• More complex training will be completed as specific questions/submissions warrant and as the
employee’s knowledge allows.


Table of Contents
Page 21 of 273
Title:
Evaluating IRB Staff Performance
Guidance Category:
Administrative
Established:
8/27/2009
Last Revision:
09/11/2023
Turnaround times are listed in business days. Study team contributions listed below are considered the
best-case scenario; the IRB cannot control additional delays on their part. Therefore, the overall “total
days” are also a best-case scenario. The IRB staff should stay within our targets each time the study team
responds to our requests for clarification or changes. Check WIRB/External IRB SOPs for TAT for those
studies.
Note: The times below should be decreased if needed due to urgency. Discuss with the Director or an
AD if you are not sure we should act on the request for urgent handling.
Note: Staff must respond to study team calls or log comments within two business days
Performance Quotient Expectations
New studies
1. Biomed new study analysts:
o 130-150 new studies per year, ~32-38 per quarter: No more than 5% PQ
2. Socio-behavioral new study analysts
o 120-150 studies, ~30-38 per quarter: No more than 6% PQ
3. Hybrid new study analysts:
o 130-150 studies, ~32-38 per quarter: between 5 and 6% PQ
Modifications
• 90-110 Modifications per quarter: less than 1% PQ
Reportable new information submissions
• 36 to 40 cases per quarter: no more than 5% PQ
Turnaround times and performance quotients are based on getting ~35 new studies per quarter, and ~100
Modifications per quarter, while reassigning all Continuing Reviews to an AA.
Variations from the above numbers due to understaffing or changes in submission volume will be
considered when evaluating performance.
For AAs processing Continuing Reviews, the targets are based on office requirements and alerting
Associate or Assistant Director if other tasks need to be adjusted in order to accomplish this.
For reportable new information submissions, these numbers will not apply if team Q is assisting the office
with other tasks or has not a full team to work on cases.
Table of Contents
Page 22 of 273
Type of Work
Initial Staff Screening
Omnibus Form
Deadline and
Pinging
Schedule
F/up ltr out
Goal
FB New
Triage upon receipt or
within 3-7 d of
assignment depending
on prioritization
Monday before
the meeting
(Friday for
Tuesday
meetings)
2 d of MTG
the aim is less than 1
calendar month or less
FB Mod
Triage upon receipt; 3-
6 d depending on
prioritization
Monday before
the weekend
(Friday for
Tuesday
meetings)
2 d of MTG
the aim is 3wks or less;
PRIORITIZE based on
relevance to subject safety
FB CR (once
45d or less
pre-exp date)
Screen no later than 3
weeks from expiration
(4 is better); earlier if
Grady study; later if
submitted less than 30
days before the
expiration
Monday before
the weekend
(Friday for
Tuesday
meetings)
2 d of MTG but
lower priority
than New and
Mod – UNLESS
Grady, expiring,
or study team
needs for other
reason
The ideal is IRB FB review at
least 2 weeks prior to
expiration, but 1 week if not
possible
FB Post-
Deferral
Resubmission
3d – have Chair weigh
in on adequacy of
response before
sending back to Full
Board
Monday before
the weekend
(Friday for
Tuesday
meetings)
2 d of MTG
Send to the same panel if not
urgent or submitted near
that meeting; if urgent
discuss with TL or Director as
to whether we can send it to
a different panel.
Post-Pending
Response
2d of receipt
2 d of final
approval
If the pending response is
acceptable, aim for <6d;
otherwise, the aim is 2wks or
less
Type of Work
Initial Staff Screening
Pinging schedule
F/up ltr out
Goal
Simple
Expedited New
Triage upon receipt or
within 3-7 d of
assignment depending
on prioritization
2 d of decision
the aim is 3 wks or less
Complex
Expedited New
Triage upon receipt or
within 3-7 d of
assignment depending
on prioritization
2 d of decision
the aim is 4 wks or less: USE
PHONE OR EMAIL to resolve
issues whenever possible to
avoid delays (log notes in
the study too)
Expedited Mod
by Staff
Triage upon receipt; 3-
5 d depending on
prioritization
same day as
approval
the aim is 1wk or less

Table of Contents
Page 23 of 273
Expedited Mod
by DR
Triage upon receipt; 3-
6 d depending on
prioritization
2 d of decision
the aim is 3wks or less
Expedited CR
(once 45d or
less pre-exp
date)
Screen no later than 3
weeks from expiration
(4 is better); earlier if
Grady study; later if
submitted less than 30
days before the
expiration
2d of decision
The aim is IRB DR review no
later than 1 week prior to
expiration (more is better)
when study team submits at
least 30 days before the
expiration
HSR
Determination
Acknowledge
immediately; 3d of
assignment to screen.
For each subsequent
response, the IRB staff
should reply within 2
days.
same day as
determination
the aim is < 1wk (*) – we do
not wish to hold up projects
that do not require any IRB
oversight.
Exempt
Triage upon receipt or
within 3-7 d of
assignment depending
on prioritization
2 d of decision
the aim is 3 wks or less
RE case: SNC
or CNC
Triage within 1 to 2
days. Sent to CoRE
within one week or
sooner if having all
required case
information (**)
If applicable, the
omnibus form
should be added
one week before
the meeting
If SCN or CNC:
Friday or
Monday after
CMTE Q. If NC
or not NC, 2 to
3 days
The aim is 4 weeks or less
RNI case: UP
Associate or Assistant
Director will log a
comment indicating
this is a potential UP
case. Send to CoRe
within 1 to 2 days
If going to Q,
one week before
the meeting. If
going to other
committees,
follow meeting
deadlines.
If the case went
to FB, one day
after meeting if
involves a
safety issue
that needs to
be addressed
with an Mod. If
not, 1 to 2 days
The aim is for 4 weeks or less
RNI case: Not a
UP, NC
Triage within 1 to 2
days. Sent to CoRE
within one week or
sooner if having all
required case
information, if
applicable (**)
N/A
If expedited: 5
days
If CoRE: 2 to 3
days
The aim is for 2 weeks or less
(*) There is often a lot of discussion with study teams so these determinations usually take longer to
review, although we should aim to stay in our targets.
(**) Considering that there is some back and forth with the study team, it is acceptable to wait a week
to send a case to CoRE. If the analyst has all the information, it is expected the case to be sent to CoRe
sooner


Table of Contents
Page 25 of 273
SOP Title: Inquiries & Complaints received about IRB submissions
SOP Category:
Administrative
Established:
7/25/2018
Last Revision:
7/25/2018
PURPOSE
The purpose of this document is to explain the review process for inquiries or complaints from study
teams about studies that were processed or are being processed by an IRB analyst.
SCOPE
The SOP applies to inquiries and complaints received by Associate or Assistant Directors, IRB Director, or
other staff regarding items handled by an IRB analyst.
RESPONSIBILITIES
• IRB analyst (IA) – receives questions/complaints or addresses them when prompted by IRB Associate
or Assistant Director or Director.
• IRB Associate or Assistant Director (ADs)- receives questions/complaints and checks with staff
managing study to submit a final response for a team when applicable. The term TL is used in this
SOP referring to ADs.
• IRB Director (ID)- receives questions/complaints and checks with Associate or Assistant Director and
staff member before providing a final response to a team when applicable
• Team Q personnel- received questions/complaints and responds only if the response indicates the
IA is following procedure. If not, it will forward questions to IA and TL.
PROCEDURE
Note: Often, the TL, Team Q personnel or ID receive calls, emails with questions or complaints about
studies being reviewed by the IRB. Even if the complaint is unfounded, we are required to provide a
response, and for that reason, the above-personnel will contact IA who owns the item, and/or their TL
(or, if unavailable, another TL) for more information. There are some points to remember during this
process:
• If there is enough information from the submission/emails to confirm that the IA has followed
current procedure and turnaround times, the complaint recipient can respond directly to the
study team, copying the IA and his/her TL
• In any response given to the study team, the IA and TL will be copied, to keep them in the loop
about this question/complaint. Copying the TL is important so that the TL can help the IA with
follow-up if any. The TL can also provide feedback to the IA if needed.
• The IRB leadership understands that mistakes will be made by IAs just because we are all human
beings. Complaints will only lead to performance feedback if there is a pattern of making the
same mistake multiple times (including not following turn-around-times) or there is a lack of
responsiveness from the IA’s part with no justification (e.g. sick or vacation leave).
If question/complaint is received by IA directly
• The IA should review the email with the question/complaint in the next two business days.
When responding to a question, the TL does not need to be included. If this is a complaint, the
IA should copy their TL.

Table of Contents
Page 26 of 273
• If the study team is emailing/calling the IA about the same issue multiple times in a two-day
period, and the review is following approved turn-around-times, the IA may reply to the study
team, copying his/her TL, letting them know that the request/question was received and that
we are working as fast as possible to resolve it.
o If there are extenuating circumstances, the TL may offer to take ownership of the
submission or other matter, for special handling and to avoid burdening the IA. This will
be the exception in cases where there is a real need, and TL should remind the study
team of this.
• If the study team is raising their voice, being unreasonable about their request, and despite
using crucial conversation tactics, the study team is not cooperative, the IA will forward their
question/concern to the TL to address.
If question/complaint is received by a Team Q personnel
• If there is enough information from the submission/emails to confirm that the IA has followed
current procedure and turnaround times, the Team Q person can respond directly to the study
team, copying the IA and his/her TL (if not self)
• If there is not enough information to show that the IA is following the procedure, the request
will be forwarded to the IA and his/her TL.
If question/complaint is received by IRB Associate or Assistant Director
• If there is enough information from the submission/emails to confirm that the IA has followed
current procedure and turnaround times, the question/complaint recipient can respond directly
to the study team, copying the IA and his/her TL (if not self)
• If there is not enough information to show that the IA is following the procedure, the TL will
copy the staff member in their response letting them know that the email was received and that
the matter is being reviewed.
• The TL will contact the IA and their TL (if not self) to look into the matter.
o If the issue was a lack of documentation, the IA will add to their record their
communication with the team. The original TL (if available – otherwise the IA or their
TL) will then answer, letting them know that the information was reviewed and that the
procedure was followed, copying all parties.
o If during the review of the question the IA’s TL and IA found that an error was made, the
IA will work with the TL on resolving the issue, and the IA will email the study team
letting them know about the correction of the human error.
If question/complaint is received by IRB Director
• If there is enough information from the submission/emails to confirm that the IA has followed
current procedure and turnaround times, the ID can respond directly to the study team, copying
the IA and his/her TL
• If there is not enough information to show that the IA is following the procedure, the request
will be forwarded to the IA and his/her TL, and the ID will email the study team back letting
them know that the email was received and that the information is being reviewed by the TL
and IA.
o If the issue was lack of documentation, the IA will add to their record their
communication with the team. The original TL (if available – otherwise the IA or their
TL) will then answer, letting them know that the information was reviewed and that the
procedure was followed. Copy all parties.

Table of Contents
Page 27 of 273
o If during the review of the question the IA’s TL and IA found that an error was made, the
IA will work with the TL on resolving the issue and the IA will email the study team
letting them know about the correction of the human error
• The TL will provide an update to the ID when the matter is resolved if ID requests.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 28 of 273
SOP Title:
IRB Staff Meeting Facilitation
SOP Category:
Administrative
Established:
02/24/2023
Last Revision:
PURPOSE
This SOP details the process of facilitating an IRB staff meeting.
PROCEDURES
1. The IRB staff member designated to facilitate an IRB staff meeting is responsible for requesting
agenda items from the IRB staff one week prior to the staff meeting and compiling them into a word
document that will serve as the agenda and eventually the recap for the meeting.
2. No later than close of business Thursday before the schedule staff meeting, save the document in
the folder located here: H:\General\IRB Org and Management\1-Staff Meeting Recaps and email it
to the Director and ADs.
3. During the meeting, record the discussion of each item in the word document and save changes.
This document will be saved for future reference for IRB staff.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 29 of 273
COLLABORATIVE RESEARCH / CENTRAL IRBS
SOP Title:
External Collaborators Included in Initial Submissions or in Modifications to
Approved Non-Exempt Research
SOP Category:
Collaborative Research / Central IRBs
Established:
9/20/2012
Last Revision:
8/15/2024
PURPOSE
The purpose of this SOP is to outline the process an analyst uses when an external collaborator is
included in an initial submission or is being added via a modification to an approved Emory study. This
SOP does not apply to exempt research or to studies where Emory has agreed to serve as the single IRB
of record for multiple enrolling sites. For new reliance agreements covering groups of studies (e.g. for a
research network), consult with the reliance AD.
For more information, please review the following guidance documents:
• OHRP Guidance on Engagement: http://www.hhs.gov/ohrp/policy/engage08.html
• OHRP Guidance on IIAs: http://www.hhs.gov/ohrp/policy/guidanceonalternativetofwa.html
DEFINITIONS
• Reliance Agreement – Documentation that an institution or individual engaged in human subjects
research has delegated institutional review board (IRB) review to an independent IRB or an IRB of
another institution.
• IAA – IRB Authorization Agreement
• IIA – Individual Investigator Agreement
RESPONSIBILITIES
• IRB Analyst (RPA) – Recognizes when another institution or individual is collaborating in research
with Emory. This is often discovered when an external collaborator is listed in the smart form or on
the first page of the protocol. The RPA communicates with the study team and the reliance team to
complete the reliance process.
• Institutional Official (IO)/IRB Director – Reviews and signs all proposed reliance agreements
(Director signs if not federally-funded or if IO is not available within a reasonable timeframe).
CONSIDERATIONS
Reliance only applies to non-exempt human subjects research. If a study has been determined to be
exempt, a reliance agreement is not applicable. External researchers that have access to an IRB, such as
those at other academic institutions, should consult with their IRB as to whether they need to submit to
their local IRB for an exempt determination. If they do not have access to an IRB, the external researcher
may remain in the smart form and Emory’s determination will be understood to cover the external
researcher.
In general, Emory is open to reviewing for another institution or individual investigator when:

Table of Contents
Page 30 of 273
• The other institution is not a separate site in a multisite clinical study (i.e. not when the other site is
enrolling and performing the protocol interventions on their own patients/subjects)
• Use of a single IRB is required by the regulations.
• The other institutions or individual investigator’s activities are minimal risk.
• Local context is not expected to be a significant factor.
• The individual investigator has no institutional affiliation and therefore no other possible IRB (note:
Community Physicians may not conduct research at EHC facilities without Emory faculty
collaborating and assuming responsibility for the study as PI.)
When in question, contact the reliance team.
PROCEDURES
For AVAMC studies, consult with IRB-VA liaison prior to proceeding.
1. Review the guidance posted on the IRB’s Collabortive Research page for adding external
collaborators.
2. If use of a single IRB is not required by the sponsor or by the regulations or the research has been
determined to be exempt, the external collaborator should obtain IRB approval/determination from
his/her IRB. Log a comment to the study team telling them this.
3. If the research is non-exempt, the external collaborator is engaged in human subjects research and
is located within the U.S., direct the study team to follow the guidance posted on the IRB’s website
on the Collaborative Research page. The study team will need to email the reliance team with the
requested information and then upload the completed reliance documents into the external team
member section of the smartform once they are completed and signed.
4. If the reliance team agrees the IRB can provide oversight to the external collaborators, confirm the
protocol describes the research activities that will be conducted by the external collaborators and
that the reliance agreement, local context review form, external team member list and engagement
determination checklist are all included in the smart form.
5.
6. Continue processing the Modification to add the external collaborator(s).
7. Once the study or modification including the external collaborator(s) is approved, log the comment
below in the study workspace and include this sentence in the approval letter “A reliance
agreement has been executed for INSTITUTION/INVESTIGATOR to rely on Emory's continued review
and oversight of this protocol.:
Dear Study Team,
A reliance agreement has been executed for INSTITUTION/INVESTIGATOR to rely on Emory's
continued review and oversight of this protocol. Please provide a copy of this approval letter to the
relying institution’s IRB.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/30/2021
Minor edits for clarity of the process, added need to approve modification and archived
changes logged over 1 year
1/12/2022
Revised process so Reliance AD handles all IAAs and IIAs.
4/28/2022
Edits for clarity


Table of Contents
Page 32 of 273
SOP Title: Obtaining Access to WIRB and NCI CIRB
SOP Category:
Collaborative Research / Central IRBs
Established:
11/8/2016
Last Revision:
01/07/2022
PURPOSE
The purpose of this SOP is to outline the steps necessary to provide IRB staff with access to accounts
with WCG IRB and NCI CIRB.
SCOPE
The SOP applies to Emory IRB staff that will be processing studies that will be reviewed by WCG IRB and
the NCI CIRB.
PROCEDURE
WCG IRB Studies: (see WCG IRB Study Processing SOP for more information)
• To gain access to the Emory WIRB Listserv: Send an email to Julie Martin asking to be added to the
WIRB listserv. Julie will log into listserv.cc.emory.edu and add the IRB staff as a subscriber to the
WIRB Listserv.
To gain access to WCG Connexus, use the login wirb-l@listserv.cc.emory.edu
and the most recent
password which can be found in the AA One Note page.
NCI CIRB Studies: (see NCI CIRB SOP for more information)
• To gain access to the NCI CIRB IRB Manager: Request a new account from NCI CIRB IRB Manager –
https://eapps-ctep.nci.nih.gov/iam/index.jsp
.
• Request that the Director or designee email [email protected] to update the roster to
reflect the IRB staff as an Institutional Contact
• To gain access to CTSU Registered Member Website: The analyst must register with the CTEP-IAM
registration system.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/30/2021
Revised WIRB instructions and archived log of updates over 1 year old
01/07/2022
Replaced Rebecca with Julie Martin as point of contact for Listserv additions

Table of Contents
Page 33 of 273
SOP Title:
WCG Listserv Duties
SOP Category:
Collaborative Research / Central IRBs
Established:
12/9/2014
Last Revision:
8/15/2024
PURPOSE
The purpose of this document is to outline the steps to address emails in the WCG Listserv.
WIRB Listserv Monitor Duties /Institutional Sign-Off
WIRB Listserv is copied on communications between WCG IRB, the study team and the Emory IRB.
Emails with “Site Pre-Review” and “Sponsor/CRO Pre-Review” do not require action by the Emory IRB
because they are requesting input from the study team or the sponsor/CRO.
1. Review the WIRB listserv emails briefly to make sure there is not an issue that we can help with.
Issues that Emory IRB needs to respond to include deviations to Emory’s language, requests for
Institutional Sign-off, study closeouts and reportable events.
• Deviations –
• Emails from WCG IRB regarding deviations from the external IRB consent checklist
should contain wording similar to “Institutional Pre-Review” or “Institutional Sign-Off” in
the subject line. Review the body of emails to determine if the Emory IRB is being asked
to provide information.
• Forward the listserv email to the owner of the study.
• The analyst assigned to the study is responsible for resolving the deviation following the
steps below.
o Review the changes being requested to Emory’s language.
o Email the reliance Assistant Director to review the changes. If changes are
minimal, include the exact text that is being changed within the body of the
email, highlighting or striking-out text to make the deviations clear. Otherwise
include a track-changes copy of the consent(s) and point out the pages that
contain the deviations.
o If the deviations are acceptable, respond to the listserv email with “These
deviations are acceptable to Emory,” and send to WCG IRB client services,
copying the study team. Save a copy of the approval email from the reliance
Assistant Director as a private comment in the study space.
o If the deviations are not acceptable, send a response to WCG IRB letting them
know the deviations are not accepted and why.
• Request for Institutional Sign-Off –
• If the request for Institutional Sign-Off relates to deviations to Emory’s language, follow
the steps above.
For other requests for institutional signoff, confirm there is a submission in eIRB for that study. If so,
confirm whether institutional signoff has been given. If not, notify the study team they are not to submit
to WCG IRB until the Emory IRB has issued signoff. Copy the analyst assigned to the study.

Table of Contents
Page 34 of 273
1. Check each regulatory document email (including “Change in Research,” “Continuing Review” and
“Closeout”) to determine if it is for an initial approval, study closeout or a change in PI. If the email is
for a closeout, pdf the email and log as a comment in the history tab of the study and close the
study.
Reportable New Information (RNIs)
Study teams are required to report local adverse events directly to WCG IRB if they meet WCG IRB’s
reporting criteria. They are also required to report events to the Emory IRB per the guidance posted on
the Emory IRB website. Once WCG IRB receives a report from a study team, WCG IRB copies the WIRB
listserv on emails to the study team and/or the Emory IRB in regard to these reports.
1. The WIRB listserv monitor reviews these emails to determine if WCG IRB is requesting information
from the IRB and notifies the Director and/or Associate Director of email communications regarding
Serious or Continuing Noncompliance, Unanticipated Problems involving Risks to Human Subjects or
Others. WCG IRB posts its reporting criteria on the WCG IRB website for study teams to reference.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
3/11/2021
Added steps for reporting continuing review data and updating approval and expiration dates at CR,
clarification about SJHC and JC site process, revised processing of deviations so the owner/AA
handles instead of the listserv monitor.
5/24/2021
Adding links to other SOPs; updating Master CT list information, minor edits
8/30/2021
Updated WIRB to WCG where applicable and added need to request WIRB approval letter and
master consent and Archived logs of updates over 1 year old
1/07/2022
Revised SOP to reflect only listserv duties, WIRB study processing now follows SOP “XIRB Study
Processing When Emory Relying on An External IRB Other than the NCI CIRB”
7/5/2022
Removed steps to email OCR and OTT, added guidance to follow cost option SOP
7/6/2023
Replaced AA
8/15/2024
Updated to current process – WCG listserv has very limited use now

Table of Contents
Page 35 of 273
SOP Title: NCI CIRB studies processing
SOP Category:
Collaborative Research / Central IRBs
Established:
5/31/16
Last Revision:
9/11/2023
PURPOSE
The purpose of this SOP is to outline the steps IRB staff uses to process studies reviewed by National
Cancer Institute’s Central IRB (CIRB), and to facilitate maintenance of the Emory investigator roster and
institutional information for CIRB.
SCOPE
The SOP applies to all studies reviewed by NCI’s CIRB with any Emory-affiliated study sites, including
CHOA. The AVAMC may also use NCI CIRB but their process may differ.
RESPONSIBILITIES
• IRB Contact (IRB Analyst): responsible for providing access to NCI CIRB study area to study teams, as
well document the study submission to CIRB in eIRB.
• Study Team – responsible to submit up to date and accurate information to Emory IRB.
List of the institutions on the CIRB roster affiliated with the Signatory Institution (Emory)-Code
Names:
1
Emory University/Winship Cancer Institute
Emory University
GA005
CIRB Component
2
Grady Health System
GA003
CIRB Affiliate
3
Saint Joseph's Hospital of Atlanta
GA011
CIRB Affiliate
4
Children's Healthcare of Atlanta – Egleston
GA035
CIRB Affiliate
5
Emory University Hospital Midtown
GA013
CIRB Component
PROCEDURE
Note: You can find all the checklists on this SOP in this folder
.
• If study teams have questions as to how they access NCI CIRB Studies, please review and refer them
to the guidance posted on the collaborative research section of our website.
• If a study team member requests to be added to the NCI CIRB roster, please forward the request to
the reliance AD or the Associate Director when the reliance AD is out of the office.
After the Study is Submitted to the Emory IRB
• Manage the ancillary reviews and complete the steps detailed in the New Study Checklist for CIRB
Studies.
• The IRB staff logs a private comment in the history tab of the study with the completed checklist.
Confirm Reliance
• Click on “Confirm Reliance”.
• Mark Yes to #3 and click OK.

Table of Contents
Page 36 of 273
Record sIRB Decision
• Click on “Record sIRB Decision”. A window will open.
o For #1 (Determination): mark “Approved”
o For #2 (Dates): Add the dates of approval as follows:
Effective Date of Study: Enter the approval date as indicated on the NCI CIRB approval letter.
This is the date that will appear on the finalized documents.
Last day of study approval period and Last day of local site approval period: In both fields,
enter the date shown on the CIRB approval letter (found on the NCI CIRB approval or
renewal letter for the overall study).
• Note, if the study team is unable to provide the overall study expiration date, check with
the reliance AD.
Leave the other date fields empty.
o Question 3 (Approval Letter from external IRB)- Upload the site-specific approval letter from
CIRB.
o Question 4 (Common Rule regulatory requirements): Click on 2018 requirements if not FDA or
DOJ regulated, or select Pre-2018.
o Question 5 - Follow the the Pre-review and Ancillary Review SOP
to mark the applicable options
for Regulatory oversight.
o Question 6 - Select options as applicable. Note that Emory does not use broad consent, so that
option should not be marked.
o Question 7 - Review the letter to see if the study was reviewed by Full Board and the risk level
determination. In general, CIRB studies are considered greater than minimal risk.
o Question 8 - Select as appropriate (usually biomedical).
o Question 9 - Select clinical trial (if applicable), Certificate of Confidentiality (because the study is
NIH funded), collaborative and multi-site, as applicable.
o Question 10 – leave blank
o Question 11 – leave blank as the ICF/Site addendum should be part of the smartform
o Question 12 - Mark yes to finalize (stamp) the documents.
o Question 13 - Mark yes and click OK.
Note: If you had to finalize documents and the system requires you to create a letter upload the NCI
CIRB approval letter and click “send letter.”
Finalizing Documents
• Click on “Finalize Documents”. A new window will open. Do not stamp the revocation form.
• Select the Site Information and HIPAA Authorization Form Addendum and assent (as applicable).
Click OK.
• View the pdf version of the documents under the “Documents” tab. Confirm the approval date is
correct on the documents.
• The study state will be “active.” (If the study was submitted as a single site study it will be listed as
External IRB.)

Table of Contents
Page 37 of 273
Processing Continuing Review
Study teams submit continuing review data as outlined on our website using the “Report Continuing
Review Data” activity for studies in the Active or External IRB state. Note that only the PI can use the
“Report Continuing Review Data” so the study coordinator might log a comment with the continuing
review information. The IRB staff can then complete the Report Continuing Review Data on behalf of the
study team.
After ensuring that all information has been reviewed, complete the following steps:
• Ask the study team if they have any study staff changes to make at this time. If yes, have the study
team update the study staff in the smart form. Once that is done, continue with next steps.
• Click “Return to Post-Review”.
• Enter the comment “Continuing Review Update.”
• Mark yes to question 3.
• Click OK.
• Click “Edit sIRB Decision”.
• For question 1 Mark “Approved”
• Do not change the initial effective date.
• Change the date shown as the “Last day of study approval period and Last day of local approval
period” to the expiration date noted on the CIRB letter.
• For question 3, upload the NCI CIRB approval letter the study team logged as a comment.
• For question 4, click on pre-2018 if not already selected.
• Skip to the last question.
• Mark yes to the last question only if the study is an old CIRB study, and there is a consent/HIPAA
document in the submission instead of a CIRB consent plus addendum. Otherwise mark No.
• Click Ok.
• Finalize documents, if needed. Stamp the consent/HIPAA document with the approval date shown
in the CIRB re-approval letter.
• Note: If you had to finalize documents and the system requires you to create a letter upload the NCI
CIRB approval letter and click “send letter.”
• Click OK.
Remember: You do not need to re-stamp any documents (NCI IRB Consent, Assent or our addendum).
Note: If the study is marked as a single site study, you will see the letter populate twice in the header for
the study (top of the study space). If the study is marked as a multi-site study, it will only appear once.
For reportable event CR summaries, follow the SOP entitled “
Reportable New Information (RNI)
Submission Notification Process for External IRB Studies” for additional steps.
CIRB Modifications
NOTE: The study team may opt to submit a Modification/Update to separate the consent from the
addendum. This will prevent the requirement of stamping consents in the future.

Table of Contents
Page 38 of 273
The following modifications require approval from NCI CIRB, but Emory does not need to review:
• Changes to the CIRB consent and a stamped addendum that do not include changes to the
addendum and HIPAA language. If the study team submits a modification with changes to the
consent, instruct them to “discard” the modification. The Emory IRB does not process Modifications
or re-stamp documents after initial approval.
• Any translated documents not already approved by NCI CIRB. If the study team has questions, direct
them to question 7 of the NCI Institution QA/QI
• Changes to locally produced advertisements/requirements. If the study team has questions, direct
them to question 8 of the NCI Institution QA/QI
The following modifications require approval from NCI CIRB and local context review by Emory IRB:
Note: If the study team is requesting the Emory IRB review documents, they must provide tracked
versions of the documents.
• Changes to PI: Request a CIRB approval letter/notice that lists the change in PI.
Note: If the study is still enrolling subjects, the site addendum may need to be updated with the
new PI’s name.
• Changes to the previously stamped addendum such as changes to HIPAA language. Compare the
language in the site addendum to the current version of Emory’s
Site-Specific Consent-HIPAA
Addendum posted on our website. If changes are needed, request clarifications. If not, click “Accept
Site Updates.”
• Changes to a consent/HIPAA authorization, for studies that still have that type of consent (vs. the
separate consent and addendum) (analyst to process): Request a CIRB approval letter that lists the
correct version of the document.
• Changes to local study sites
• Updates requiring that will require a new ancillary review (e.g.; Procedures involving radiation,
cancer related aims added, new conflict of interest, etc.))
Modifications that do not require a Modification in eIRB, but do require re-stamping:
• For studies with a combined consent/HIPAA document: Changes to the overall study that do not
include a consent revision. For example, a protocol update will require an update to the consent
form just to update the version date to match the revised protocol. If this is the case, the study
team must submit a modification or click Update Study. Return the study to post-review to stamp
the documents with today’s date. (see below for more information).
Processing CIRB Modifications
Depending on the study status (external vs active), the study team will have the option to submit either
a modification or an update to their study.

Table of Contents
Page 39 of 273
If the study team submitted a modification
• Under the modification space, click on “View Modification.”
• Review the initial submission to see what documents are being added or modified. Use the
“history” and “compare” functions to see the differences in the documents.
• If all of the information appears to be correct, click on Accept Site Updates.
• Click “Record sIRB Decision”.
• A new window will open.
• Confirm the approval/expiration dates did not change from the initial or last CR approval by CIRB.
• Click “Approved.”

Table of Contents
Page 40 of 273
• Mark that you are ready to submit (see below)
• Click OK
• Click on “Finalize documents”
• Select the addendum to be re-stamped
• Click OK.
If the study team updated the study
If the study team does not have the option of submitting a modification, they will need to use “update
study” option. The study team may reach out to you if they do not see the “finalize update” option.
Confirm everything looks correct and click “Finalize Update”.
• Click “Return to post review”
• Mark yes to question 3
• Click on “edit sIRB Decision”.
• Click “Approved” for question 1.
• If you don’t need to finalize documents, you don’t have to change approval/expiration dates. If you
need to finalize documents, change the effective approval date to today’s date. Make sure that the
expiration date matches the current approval period.
• Upload the letter the team logged as a comment under question 3
• Click on pre-2018 under question 4 (if not selected already)
• Skip questions 5 to 11.
• Say yes to question 12 if you need to finalize documents. If no, answer “no” to this question.
• Click OK
• Finalize documents, if needed.
• Note: If you had to finalize documents and the system requires you to create a letter upload the NCI
CIRB approval letter and click “send letter.”
Processing Reportable new information submissions
Follow the SOP entitled “Reportable New Information (RNI) Submission Notification Process for External
IRB Studies” for additional steps.
Processing Close-Outs

Table of Contents
Page 41 of 273
PIs are responsible for submitting a Study Closure to NCI CIRB via IRBManager. The PI logs a comment in
the submission with a letter from the NCI CIRB indicating the study has been closed. Click “close site”.
REFERENCE
• NCI CIRB SOPs: https://www.ncicirb.org/about-cirb/sops
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
9/11/2023
Clarified CIRB processing Archiving logs older than one year
Table of Contents
Page 42 of 273
SOP Title:
Reportable New Information (RNI) Submission Notification Process for External IRB
Studies
SOP Category:
Collaborative Research / Central IRBs
Established:
12/9/15
Last Revision:
07/6/2023
PURPOSE
This SOP details the process for reviewing RNIs reported to the Emory IRB for studies reviewed by
external IRBs. This SOP also includes the process for the review of the CR at the time of the study
renewal.
TIMEFRAME
Once the Emory IRB is notified of an RNI for an external IRB study, other than in summary format at
Continuing Review, the Emory IRB aims for 2 business days to review it and forward it to the
appropriate Emory officials as detailed below. Once the RNI determination letter from the external IRB
is received, the Emory IRB must complete the procedure described below within 5 business days.
For CR summaries, contact team Q if the study team is reporting an egregious event that was not
submitted promptly during the last approval period.
BACKGROUND
• Emory study teams are responsible for submitting RNIs to the external IRB as required by the
external IRB’s reporting policies but should follow our reporting requirements if the external IRB’s
requirements are less stringent.
o If these events are determined to represent an unanticipated problem, serious noncompliance,
or continuing noncompliance the external IRB will forward the determination letter to the
Emory IRB.
• The study team is also responsible for reporting to the Emory IRB for studies reviewed by external
IRBs when the following internal events occur:
Event Type
Reporting Timeline
Egregious events
• Wrong side surgery
• Wrong drug
• Wrong patient
• Fabrication of data
• Falsification of data
• Breach of confidentiality (HIPAA Privacy Breach)
• Study Suspension (related to compliance concerns)
•
Internal related death
Promptly with an RNI
Unanticipated Problem that
•
Is an Internal event
In summary format at Continuing
Review

Table of Contents
Page 43 of 273
• Is unexpected
• Is related or possibly related to the research
• Suggests the research poses a greater risk of harm than
was previously known
• Does not fall under “Egregious events” above
Noncompliance
• Other failure to comply with laws, regulations, or
Emory policies and procedures
In summary format at Continuing
Review
PROCEDURE FOR SUMMARY AT CR
• The study team should submit via once the external IRB has reapproved the
study. The report should include a completed CR workbook
.
o If the study team is including an egregious event in the workbook, ensure this was reported
to the Emory IRB via an RNI in the last approval period. If the study did not report it to us
using an RNI, please contact team Q for the next steps.
PROCEDURE FOR RNIs
Egregious Events
1. Upon receipt of the RNI, the QA Director/Team Lead reviews it to determine if it falls into one of the
egregious events listed in the chart above. If the event is not an egregious event, the QA
Director/Team Lead will assign themselves as coordinator and administratively discard the event
with a logged comment to the team.
2. If it falls into one of the above categories, the QA Director/Team Lead will email the reliance lead to
let them know and will delegate the case to a member of Team Q for Institutional notification.
a. Complete the internal notification as noted below.
b. The team Q member then creates a file under on the H: drive in the QA Working Files\NC UP
Complaints\RE Notifications from External IRBs folder with study number and PI name,
using the following format (1234 Jones) and save all documentation related to the RNI.
c. Enter the information of the reported event in this spreadsheet
located at OneDrive/IRB-
Staff/Documents/H drive/General
3. The external IRB should send notice of its determination and the determination letter to the Emory
IRB. Request the Emory researcher to upload the determination letter in the RNI via logged
comment if it is not provided at initial submission.
4. Once all reporting is complete and any fed letters have been filed in the QA folders, the Q team
member should update the spreadsheet and should self-review the RNI submission. The Q team
member should not check any determination boxes, since these determinations were not made by
our IRB. The Q team member should instead enter the reviewing IRB’s determination in the Notes
field and submit the review. The final state is “acknowledged.” There is no need to write a letter.
4. Follow the post determination notification procedure noted below.
5. Once the external IRB determination is in, follow the ‘external IRB determination’ notification process
below for SNC, CNC, UP, suspension as applicable.
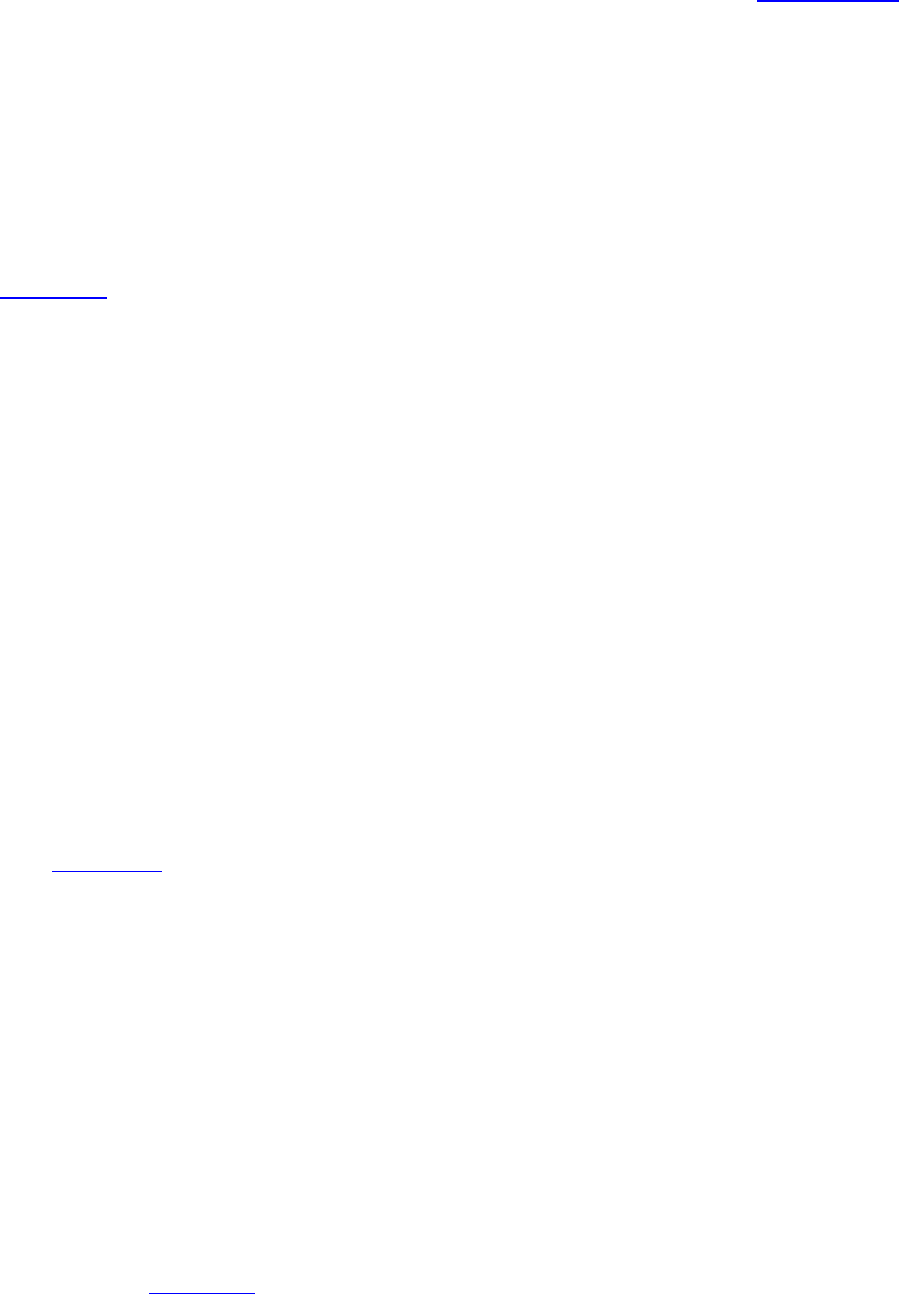
Table of Contents
Page 44 of 273
Institutional Notification (pre-IRB-determination)
While awaiting a determination from the reviewing IRB, a member of Team Q will draft an email to the
appropriate parties and will copy and paste the lay summary into the body of the email. They will attach
a pdf copy of the RNI report and any relevant attachments. Include a link to the RNI submission in the
body of the email as well.
See contact grid
for who to copy in the notifications.
PROCEDURES FOR REPORTS RECEIVED DIRECTLY FROM EXTERNAL IRBs
1. For external IRB determinations of serious noncompliance, continuing noncompliance or
unanticipated problems received directly from external IRBs, the Emory IRB person who was notified
will forward the email to the AVP for the HRPP, Reliance AD and Team Q with the information with
the RNI number (if applicable).
a. Upload a copy of the letter under the RNI submission (as applicable).
b. If an RNI does not exist, Team Q will review the information to determine if the
submission was needed (in case it was not submitted).
c. Follow the procedure under “Notification to Internal IOs” as appropriate. These
notifications are only required for determinations of SNC, CNC, or internal (at Emory
sites) UPs.
2. For external IRB determinations that an event was not SNC/CNC/internal UP, proceed to EGREGIOUS
EVENT RNI CLOSURE IN EIRB.
PROCEDURE AFTER DETERMINATION IS MADE
IRB DETERMINATION NOTIFICATION TO INTERNAL IOs
For egregious events or events the external IRB determined to be SNC, CNC, UP, update the External IRB
reporting spreadsheet
. The Q team member drafts an email to the Emory PI with the IRB # and Study
Short Title in the subject line stating the following:
i. Dear Dr. _________:
We have received notification that the [Reviewing IRB] determined that a reportable
new information submission during the course of the above-referenced study constitutes
a [type of determination]. Attached, please find the determination letter from
[Reviewing IRB]. This determination may be reported to the sponsor and federal
oversight agencies, as applicable.
It is imperative that you follow your CAPA Plan as written. If you find that there are any
inaccuracies in this letter, please notify your point of contact at [Reviewing IRB].
Please let me know if you have any other questions. Thank you for your cooperation.
2. CC based on the contact grid
Once sent, save a copy of the determination letter and the
email in the study folder under QA Working Files.

Table of Contents
Page 45 of 273
3. Note: If you receive copies of external reports from agencies or notices of receipt of external
reports from agencies, scan and/or save them under QA Working Files in the appropriate
study folder. If the report produces any results, save the contents in the same manner, and
email it to the IOs using the same process described above.
4. Save letters to federal agencies in the folder in QA working files, but there is no need to
send the letters to the IOs.
EGREGIOUS EVENT RNI CLOSURE IN EIRB
• Once all reporting is complete and any fed letters have been filed in the QA folders, the Q team
member should update the spreadsheet and should self-review the RNI submission. The Q team
member should not check any determination boxes, since these determinations were not made
by our IRB. The Q team member should instead enter the reviewing IRB’s determination in the
Notes field and submit the review. The final state is “acknowledged.” There is no need to write
a letter.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
02/24/2023
Revised IO notification letter in reference to the other notifications, linked out to
contact grid
07/06/2023
Added clarification around study suspension and archived log older than one year

Table of Contents
Page 46 of 273
SOP Title: Vetting NIH Single IRB Plans
SOP Category:
Collaborative Research / Central IRBs
Established:
5/15/2018
Last Revision:
8/21/2020
PURPOSE
This SOP outlines the process the reliance team uses to review Single IRB plans for federally-funded
multi-site research at the grant application stage.
PROCEDURES
1. Upon receipt of a request to review a single IRB plan, review the information provided by the
investigator and ask for additional information if needed. The information that is needed is the
research related activities that will take place at each site and the number and location of sites that
will participate in the research. Note: Emory is not currently resourced to serve as the Single IRB for
studies involving more than 5 sites in the U.S. and will not provide oversight to international sites.
2. The Single IRB plan may state that Emory will serve as the Single IRB, that another IRB will serve as
the Single IRB, or that the Single IRB has not yet been determined.
3. Consult with an AD or Director to determine whether Emory can serve as the Single IRB.
4. If Emory IRB is willing to be the IRB of record, notify the investigator they may proceed with the
Single IRB Plan naming Emory IRB as the sIRB.
a. Provide the Emory investigator with a Letter of Support to include with the sIRB Plan. Use
the template letter located in this folder: H:\External IRB Relationships 6.3.2020\07.
Templates
5. If Emory IRB is not willing to be the IRB of record, notify the investigator and provide two options:
a. The investigator can contact the IRBs of other participating sites to see if they are willing to
serve as the reviewing IRB. Suggest to the investigator that the IRB will need to be AAHRPP
accredited and have experience serving as the reviewing IRB for multi-site research. Instruct
the investigator to confirm whether or not the IRB charges a fee to serve as the sIRB so the
anticipated expense can be included with the budget.
b. The investigator can request a quote from WIRB or another commercial IRB and include that
in the budget submitted with the grant. Refer the investigator to the information on our
website including the quote request form.
6. Once a single IRB is identified, the investigator may use the templates on the Emory IRB website to
draft the Single IRB Plan.
7. If Emory is not serving as the Single IRB, remind the investigator to use the Single IRB Quote Request
Form found on the Emory IRB website to request a quote from WIRB and include in the budget
submitted with the grant.
8. Save the email correspondence and any letters of support on the H Drive at H:\External IRB
Relationships 6.3.2020\04. Single IRB Plan Requests. Create a new folder and name it as follows:
Last Name, First Name – Title.
LOG OF SIGNIFICANT CHANGES


Table of Contents
Page 48 of 273
SOP Title:
XIRB Study Processing When Emory Relying on An External IRB Other than the NCI
CIRB
SOP Category:
Collaborative Research / Central IRBs
Established:
1/3/2018
Last Revision:
7/5/2022
PURPOSE
The purpose of this SOP is to outline the steps IRB staff use to process submissions when Emory is
relying on an external IRB (other than the NCI CIRB).
SCOPE
This SOP applies to any multi-site study where Emory is relying on an external IRB other than the NCI
CIRB. It outlines steps for processing initial submissions as well as follow-on submissions.
PROCEDURES
1. For WCG studies, create a new folder to save a copy of the study checklist for the study here:
(H:\External IRB Relationships 6.3.2020\03. Current Umbrella IAAs\WIRB\WIRB Study Documents).
2. For other studies, create a new folder for the external IRB checklist and reliance documents:
• Go to this folder: H:\External IRB Relationships 6.3.2020\01. Current IAAs
• Right click and copy the folder “USE THIS TO CREATE NEW FOLDERS”
• Double click on the “Emory Relying” folder
• Paste the “USE THIS TO CREATE NEW FOLDERS” folder and then rename it with the following
naming convention:<initials of reviewing IRB or short name>_PI Last Name (short title of
protocol) STUDY ID
Examples:
o Mayo_Cohen (LEO) 2973
o JHM_Smith (ROP3) 4567_
3. Rename the study checklist to include the study number.
4. Review smart form for completeness. If the following are not included, withdraw the submission
and log a comment to the study team letting them know what is needed and to resubmit once the
following information is available (see template text in AA OneNote page):
• For WCG and Advarra – Completed Form A
• For other external IRB studies – Supplement to protocol
• EPEX number (if funded)
• Review the SOP titled “Cost Option for Clinical Trial Agreements and ICFs” to determine the
correct cost option.
• Ancillary review approvals (PRMC, EHSO Radiation Safety, EHSO Biosafety, COI, etc.)
• Current CITI training for all study team members

Table of Contents
Page 49 of 273
5. If the short study title does not begin with “XIRB:” and the name or abbreviation of reviewing IRB,
request that it be added. (Examples: XIRB: Advarra, XIRB: UNC, XIRB: WUSTL, XIRB: Utah) Studies
going to WCG may just begin with “WIRB” or “WCG.”
6. Use the XIRB study checklist to review external IRB studies. Complete the checklist and attach to a
logged comment listing pending items so the study team is aware of what is needed before we can
issue institutional signoff. Repost an update weekly.
7. Manage any required ancillary reviews as applicable. Refer to the SOP “Pre-Review options and
Ancillary Review Information.” You will need to update the review to “accept” it and upload the
approval document provided by the study team.
8. Confirm that all required training is current for all Emory study team members. If unsure what is
required, review guidelines posted on our website.
9. If the study team has requested Sensitive Study Status -selecting Yes to Q5 in For Clinical
Research/Expanded Access Only:
• Manage Ancillary Reviews
• Assign the review to Dr. Gunthel – Do not select a department or a review type.
• The response is required
• E-mail Dr. Gunthel with a study link, study number, and an explanation of the review. Please
request Dr. Gunthel log a comment so that it shows in the study history.
10. If applicable, ensure that Margaret Huber in the Research Integrity and Compliance
(mhube[email protected]) has completed a REMS (REMS mentioned in protocol) review of the study, or
Controlled Substance form (box is marked in smart form). See new study checklist for more
information.
11. If changes to the smart form or uploaded documents are needed, click “Request Pre-Review
Clarifications” and describe the changes that are needed.
12. If provided, complete the Local Context Review form provided by the reviewing IRB. If the form
requests specific information regarding the study activities that will be conducted by Emory, forward
the document to the Emory study team for completion of their portion of the form. Review
completed form with reliance AD. If a LCR form is not provided in the smart form, use the Emory
template found in this folder: H:\External IRB Relationships 6.3.2020\06. Forms and Checklists.
13. Once the local context review process is complete, confirm with the Reliance AD if there is a fully
executed reliance agreement in place (there is a master agreement in place with Advarra and WCG
IRBs) and click on the Confirm Reliance button on the left side of the study workspace in eIRB.
14. Upload the executed reliance document (N/A for WCG or Advarra), the completed local context
form, completed external IRB consent checklist and COI management plan (if applicable).
15. Click the Yes button to confirm reliance on the single IRB of record. Click OK. The study should now
be in the Pending sIRB Review state. (Once the site-specific approval letter and ICFs are available
from the reviewing IRB, the study team is responsible for providing these documents to the Emory
IRB via a logged comment in the smart form.)
16. Review the external IRB approval letter to confirm it is for the correct study and PI and is a full
approval. Discuss with the reliance AD if you have questions about the letter.
17. Click on Record sIRB Decision on left of study workspace.
18. Select the correct determination. (This should be “Approved.”)
19. Enter the initial approval date of the study and the initial approval date of the site. Enter the last day
of study approval period and the last day of local site approval period.

Table of Contents
Page 50 of 273
20. Upload the approval letter from the reviewing IRB.
21. Mark the appropriate options for the remaining questions 4-9.
22. Mark No to Finalize Documents.
23. Mark Yes to record decision and click OK.
24. The study should now be in an Active state.
25. Confirm the dates are correct under the word Active on the top left corner of the main study page. If
the dates are not correct, return the study to post-review.
26. Click on Edit sIRB Decision and enter the correct dates. Click OK. The study should appear back in the
active state.
27. Log this comment to the study team “Emory has acknowledged the approval letter from the
reviewing IRB and your study has been moved to an Active state. Refer to the Collaborative
Research page on the Emory IRB website regarding study team responsibilities for submitting
continuing reviews, modifications, and reportable events when Emory is relying on an external IRB.”
Beyond Initial Approval:
Modifications:
1. Study teams are responsible for notifying the Emory IRB of certain changes to ongoing research as
noted on the collaborative research section of the Emory IRB website. Depending on the study
status (“Active” vs “External IRB”), the study team may report changes by submitting a modification
(that we approve) or will use the “update study” function (that we accept). We prefer study teams
to use the modification function if they have it. If they do not have that option, the study team will
need to use the update study activity. Be aware that study teams are able to accept site updates
themselves. They should not do this, and AAs are still responsible for reviewing updates.
Review the external IRB approval letter and confirm the changes being reported to the Emory IRB
match what is noted in the approval letter.
2. For modifications to add or remove Emory study personnel, follow the SOP “Adding/Removing Study
Personnel.” AAs can approve these modifications.
3. For modifications indicating increased risk, follow the SOP titled “Modifications: Mods indicating
increased risk.”
4. For modifications that require new ancillary reviews such as a new COI, change to radiation
procedures, infectious agents, etc., consult an Assistant Director for guidance.
5. For S-I Studies Only:
• If study team submits a modification that includes addition of external sites or identifies
new risks, trigger a new S-I ancillary review so that Margaret Huber receives notification.
• If the study team is requesting to add external sties, log this comment to the study team:
“If you plan to add participating external sites, contact the reliance team lead at
irb.reliance@emory.edu. In addition, you are required to notify the S-I advisor before
external sites are activated by sending her a completed S-I IND Responsibilities Checklist
Multi-site Trials for review / S-I IDE Responsibilities Checklist Multi-site Trials for review.”
• The modification cannot be approved without S-I ancillary signoff.

Table of Contents
Page 51 of 273
Changes Reported using Update Study activity
6. When the study team uses the “update study” function, the owner/AA receives an email. Review
each update.
7. When updates are finalized, the owner/AA will receive an email “Emory eIRB Notification of Finalize
Updates.” The owner/AA needs to look at each one to confirm the study team has not finalized the
update themselves.
8. Review the changes reported and finalize the update. The owner/AA can approve modifications for
study staff changes.
9. Click “Accept Site Updates”
Continuing Reviews
For S-I studies only:
• Once a month the Reliance Team Lead will run a report of XIRB studies where the Emory PI
holds the IND or IDE that shows studies expiring within 60 days.
• The Reliance Team Lead will log the following comment to the study team and trigger a new S-I
ancillary review. Insert the date of expiration and the date of the deadline to complete the S-I
review which is 30 days prior to study expiration:
“This study is due to expire on MM/DD/YYYY. As the Sponsor-Investigator of this study, you
are required to complete the IND Responsibilities Checklist Continuing Review Update / IDE
Responsibilities Checklist Continuing Review Update for this study at each continuing
renewal if subjects remain in follow-up and send to the S-I advisor (Margaret Huber,
mhuber@emory.edu) for review. You must complete the S-I review at Emory by
MM/DD/YYY”
• Continue processing as below.
Begin here for Studies that are not S-I studies:
• Study teams are expected to report CR data to the Emory IRB. The report should be made upon
receipt of the CR approval letter provided by external IRB. If the PI is not available to report the
data, it may come in via logged comment.
• If the data comes via logged comment, confirm that the IRB renewal letter is for the correct study
and the CR Workbook is attached.
• If relevant documents are attached, click “Report Continuing Review data” on the left menu. A new
window will open.
• In #4, upload the IRB approval letter and CR Workbook and click OK.

Table of Contents
Page 52 of 273
• Click “Return to Post-Review” on left menu.
• Click the box to the right of question #3.
• Click OK.
• Click “Edit sIRB Decision.” Do not change initial approval or initial effective dates.
• Enter the expiration date noted on the approval letter in the fields for the “Last day of study
approval” and “last day of local site approval.”
• Upload the CR approval letter in #3.
• Click OK.
• Confirm the dates entered are correct on the main study page. If not, click “Return to Post-
Review” and complete the process again.
• The study team is also expected to update their study personnel list at this time. Specifically,
they need to add any team members that were added during the prior approval period. If you
do not see a corresponding update with the report of CR data, please request. Once the
personnel are added, training should be confirmed per the SOP entitled “Training Verification.”

Table of Contents
Page 53 of 273
Reportable New Information (RNIs)
For S-I studies Only:
1. Notify Reliance Team Lead upon receipt of RNI.
2. Trigger S-I ancillary review for the RNI and notify Q.
3. If S-I review determines there is an issue that should be escalated to the external IRB, the S-I
Reviewer will notify the Reliance Team Lead and Team Q lead within two business days if egregious
event and within five business days if not egregious event.
4. The Reliance Team Lead or Team Q Lead if Reliance Team Lead unavailable, will notify the reviewing
IRB point of contact within two business days.
5. Upon receipt of response from reviewing IRB point of contact, Reliance Team Lead or Team Q lead if
Reliance Team Lead unavailable, will log a private comment in the history of the study with the
correspondence.
6. Reliance Team Lead or Team Q lead if Reliance Team Lead unavailable will follow up with the
reviewing IRB point of contact weekly until the matter is considered closed at the reviewing IRB.
For All Studies:
Study teams are required to report local adverse events directly to the external IRB if according to the
external IRB’s reporting criteria. They are also required to report events to the Emory IRB per the
guidance posted on the Emory IRB website.
Save all emails related to RNIs and Unanticipated Problems under the WCG IRB Study Document folder
(H:\General\External IRB Relationships 6.3.2020\WIRB\WIRB Study Documents W00000XXX) after
notifying the Q team.
Follow the SOP entitled “
Reportable New Information (RNI) Submission Notification Process for External
IRB Studies” for additional steps.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/24/2021
Adding additional information to the checklist process
8/30/2021
Updated process and archived log of changes over 1 year old
9/20/2021
Revised to make applicable to Advarra studies, removed detailed steps that are
covered in the checklist
01/07/2022
Revised to reflect new process, include WCG studies, and provide steps for
follow on submissions
04/6/2022
Added guidance for S-I studies
4/22/2022
Archived Updates over 1 year old in log of changes
4/28/2022
Minor edits to remove specific logged comment and specific criteria for mods
7/5/2022
Added guidance to follow cost option SOP

Table of Contents
Page 54 of 273
SOP Title:
XIRB Study Processing for Studies Transitioning from Emory IRB Approval to
External IRB Review
SOP Category:
Collaborative Research / Central IRBs
Established:
8/21/2020
Last Revision:
02/24/2023
PURPOSE
This SOP outlines how the IRB Analyst/Reliance RPA will process studies transitioning from Emory IRB to
External IRB review when required by federal regulations.
SCOPE
This SOP applies to any study currently approved by the Emory IRB that is required to use an sIRB due to
the NIH Single IRB Mandate or the Cooperative Research Component of the Revised Common Rule.
PROCEDURE
1. Instruct the study team to submit an external IRB study in eIRB and include the following
documents:
• Emory original IRB approval letter
• all current documents from the previous IRB smart form
• study-wide approval letter from the sIRB
• local context review form and reliance document if provided by the sIRB
2. Instruct the study team to log the following comment when they submit the XIRB study.
“This is a resubmission of Emory IRB00xxxxxx. This study is now required to transition to the [NAME]
IRB that will serve as the single IRB of record for this study. Per the Emory IRB, department review
and other ancillary reviews do not need to be repeated.” If the study team did not log this comment,
the analyst will need to do it.
3. Follow the SOP titled “XIRB Study Processing When Emory Relying on An External IRB Other than the
NCI CIRB.”
4. Email OCR and OSP, copying the PI and study coordinator that the IRB number for the project has
changed from the original Emory IRB number to the new external IRB study number.
5. Instruct study team to close out original Emory-approved study.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
3/11/2021
Added notification to OCR and OSP when IRB number for study changes
8/30/2021
Minor edits for clarity and archiving logs of changes over 1 year old
4/28/2022
Simplified to refer to SOP
02/24/2023
Removed reference to reliance request form which we no longer use.

Table of Contents
Page 55 of 273
SOP Title: Closing Out Multi-Site Studies with Central or Single IRB Review
SOP Category:
Collaborative Research / Central IRBs
Established:
3/30/2018
Last Revision:
3/11/2021
PURPOSE
This SOP details how to close out studies that are multi-site 1) when Emory has ceded review to an
external IRB and 2) when Emory is the IRB of record for external sites.
RESPONSIBILITIES
• Analyst – processing the close-out from the IRB side for studies where Emory is the IRB of
record.
• Reliance RPA- processing the close-out from the IRB side for studies where Emory is relying on
another IRB.
• Emory study team- collecting all necessary information and documents for the close-out and
initiating the close-out. Being a liaison between the IRB and the other study teams.
PROCEDURE
For External IRB Studies:
1. The study team submits a “Report Continuing Review Data” and uploads the closeout letter from
the external IRB.
2. Click on “Close Site” from left menu.
3. This activity will discard all pending modifications but pending RNIs will remain. Review the pending
modifications that appear in the window before closing the study.
4. Enter a comment and click OK.
5. The study is now closed.
Where Emory is the IRB of Record:
Site Closure:
1. To close a site while the rest of the study remains open, the study team submits “Report
Continuing Review Information.” The study team provides a study summary detailing why the
site is closing and providing the information requested in the activity.
2. Click “Close Site” from left menu.
3. Log a close out comment using the template language document on the H drive and click OK.
The site is now in a closed state.
4. Click Prepare Letter from left menu. There is no option for a site closure letter, so use the
Closure Template letter saved here on the H drive External IRB Relationships Folder 6.3.2020
Folder> 07. Templates.
5. Upload revision.
6. Name the letter “STUDYXXXXXXXXX_Site Number_Site Name_Closure_date” and click OK.
7. Click “Send Letter” from left menu.
8. Click OK.
Study Closure:

Table of Contents
Page 56 of 273
1. The Emory study team clicks on Sites tab, selects a site, and reports continuing review
information. They will do this for each active site. The study team marks “Yes” to “Report
Completed” for each site and clicks “Submit” on the left menu.
2. The Emory study team submits a Continuing Review indicating they want to close the study and
completes the fields.
3. Review the CR.
4. Complete the pre-review.
5. Assign to a designated reviewer.
6. The designated reviewer completes their review.
7. The study will be in post-review.
8. All pSites, CRs ad Mods must be closed before the study can be closed. Click on Sites tab and
select a site.
9. Click “Close Site” from left menu, enter a comment and click OK.
10. Click Prepare Letter.
11. Select the PI Study Closure Letter and edit as needed.
12. Upload revision
13. Name the letter “STUDYXXXXXXXXX_Closure_date” and click OK.
14. Click Send Letter. The CR is now approved, and the study is closed.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/31/2019
Replaced “reliance specialist” with “reliance RPA”
9/7/2020
Updated process for SaaS

Table of Contents
Page 57 of 273
SOP Title: Issuing Partial HIPAA Waivers for XIRB Studies
SOP Category:
Collaborative Research / Central IRBs
Established:
2/1/2019
Last Revision:
1/04/2023
PURPOSE AND SCOPE
This SOP outlines the process for the Emory IRB to issue a partial HIPAA waiver for recruitment
purposes to Emory study teams when Emory has ceded IRB review to an external IRB and that
IRB does not issue such waivers. This SOP only applies when Emory has been designated as the
Privacy Board for the Emory study team under the reliance agreement.
Note: WCG IRB and Advarra IRB both serve as the Privacy Board as do most academic IRBs.
PROCEDURE
1. Ensure the study team has completed the HIPAA Applicability and Waivers Requested
section of the smart form and uploaded the HIPAA Applicability and Waiver Worksheet.
2. Review the information provided by the study team and request clarification if needed.
3. Send an email to the Emory IRB Asst or Associate Director requesting the partial HIPAA
waiver for recruitment purposes. The IRB number should be included in the subject line of
the email.
4. The authorized individual should review the waiver request within five (5) business days. (If
not, send a reminder email.)
5. The authorized individual will log a comment in the history tab in eIRB indicating if the
waiver has been granted.
6. Include the determination regarding the partial HIPAA waiver in the institutional sign off
and if applicable, the local context review form that Emory completes for the reviewing IRB.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
4/11/2019
Updates to follow current process
8/31/2019
Replaced “reliance specialist” with “reliance RPA”
8/21/2020
Updated for current process
1/20/2021
Updated to provide more details
4/28/2022
Revised SOP title and added clarification about scope.
7/05/2022
Added Advarra

Table of Contents
Page 58 of 273
SOP Title:
Processing New Studies When Emory is the Single IRB of Record (sIRB) for a Multi-
Site Study
SOP Category:
Collaborative Research / Central IRBs
Established:
8/21/2020
Last Revision:
02/24/2023
PURPOSE
This SOP outlines the steps to process studies when Emory IRB is serving as the sIRB.
SCOPE
This SOP applies to any new multi-site study for which Emory has agreed to serve as the sIRB for other
sites that will enroll participants. (For studies with external collaborators that will not enroll participants
but who wish to rely on Emory IRB for review, see the SOP titled “Analyst Process of IRB Authorization
Agreements (IAA) or Individual Investigator Agreements (IIA) Reliance Agreements When Emory is the
Reviewing IRB for External Collaborators.”)
Note: If the study is an S-I study, consult the Assistant Director for the reliance team before proceeding.
PROCEDURE
1. Upon confirmation that Emory will serve as the sIRB for the study, the reliance analyst will instruct
the study team to submit a multi-site sIRB study in eIRB.
a. Instruct the study team to insert “sIRB - ” prior to the short study title.
b. If the study is also an S-I study, instruct the study team to insert “S-I” after the short study
title.
2. If more than one relying site will enroll participants, the Emory study team’s project manager/lead
coordinator will set up a study in the IRB Reliance Exchange (IREx) by logging into the website at
https://www.irbexchange.org/p/
. Directions provided below to assist if needed.
a. Create a Study
i. Provide description of the study
ii. Enter the protocol date/version
iii. Upload the protocol and mark as Draft. It is important to mark as Draft so the
protocol can be updated later.
iv. The Review & Submit screen as shown below will appear. Confirm the protocol is
marked as a draft in red. If all information is accurate, click Save at the bottom.

Table of Contents
Page 59 of 273
v. Complete the Study Setup. Mark Yes for the first two questions, mark that sites will
provide the consents for sIRB review, and mark No for LOI. (LOI will be included in
the SMART IRB LOA if applicable for each site.)
b. Identify the study manager. Confirm the study manager with the PI if it is not clear. The
study manager will manage the flow of documents between the Emory IRB and sites.
Study Processing Steps:
1. Once the study is submitted in eIRB and assigned to a reliance analyst, the reliance analyst will
follow the SOP titled “eIRB Processing of New Study Applications – Preliminary Analysis Through
Approval.” If the study is an S-I study it will be assigned to an S-I analyst.
Note: The initial approval will be for the protocol and master consent form that will be distributed
to the external sites as well as the approval for the Emory site.
a. Ensure the study team uploads all master consent forms, recruitment materials, and any
other study-wide documents in the Study-Related Documents section of the smart form.

Table of Contents
Page 60 of 273
Enrolling sites will use the site addendum to add their institutional language to the approved
master consent form later in this process.
b. Ensure the Emory site-specific consent form and recruitment materials are uploaded in the
Local Site Documents section of the smart form.
c. Finalize only the Emory site-specific consent form and not the master consent form.
d. List the master consent form(s) in the list of approved documents in the approval letter.
2. The reliance analyst will upload the approved documents in IREx.
3. After the study is in an approved state, it will appear in the study team’s External IRB tab in eIRB
with an “Active” state.
Adding Participating Sites:
1. The reliance analyst will submit a ticket to ORA-IT requesting the external site PIs be added to the
eIACUC user database. Provide them with the last name, first name, email address, and name of the
PI’s institution. The person is usually added within two business days.
2. While awaiting addition of the pSite PIs to eIRB, confirm whether the relying site institutions are
signed on to the SMART IRB Master Reliance Agreement by searching participating institutions on
the Smart IRB website.
3. Prepare the reliance instructions using the IREx template located in this folder: H:\External IRB
Relationships 6.3.2020\10. Emory as sIRB Materials\IREx Materials. If the relying institution is not
part of SMART IRB, use the standard reliance agreement template for that site. Prepare the
template reliance documents with the study-specific information.
4. Complete the study-specific information in the reliance instructions template for IREx.
5. The reliance analyst will send the following materials to the Emory study team point of contact for
distribution to relying sites:
i. Reliance instructions form for IREx
ii. Emory IRB approval letter
iii. Emory IRB approved protocol
iv. Emory IRB approved master consent form
v. SMART IRB LOA (and/or standard reliance agreement if an institution is not part of
SMART IRB), can also just use documentation in IREx system
a. In the email to the study team point of contact, instruct the point of contact to send the
materials to the site PIs, copying both the relying IRB point of contact for the site(s) and the
Emory IRB reliance email address.
b. Remind the study team point of contact that they will need to follow up on the collection of
documents until they are completed and upload them in the pSite study workspace once
completed.
c. Let the study team know that you will be creating the sites while waiting on the study teams to
complete the documents.
6. Once the reliance analyst receives confirmation that the external PIs have been added in the eIACUC
database, the reliance analyst will click “Add Participating Sites” from left menu in the study

Table of Contents
Page 61 of 273
workspace and follow the prompts to add the participating sites. The sites automatically appear
under the “Sites” tab with the state “Invitation Pending.”
7. Select Submit Invitation Decision and complete the questions. This moves the site into the state
“Awaiting Site Materials.”
8. Once the documents are completed for a pSite (reliance agreement, local context review form, site-
specific consent form) instruct the Emory study team to select the site(s) under the Sites tab and
complete the smart form for each site.
9. Confirm the study team has uploaded clean and tracked versions of the draft site-specific consent
form, in the Local Site Documents section along with site-specific recruitment materials, executed
reliance agreement, and signed local context review form or pdf copy of local context review from
IREx.
10. Once the smart form is complete and the PI clicks “Finish”, they must click “Submit Site Materials”
from the left menu.
11. Review the smart form for completeness and send back to study team if edits are needed.
12. Once the smart form is complete, click “Assign Designated Reviewer” from left menu and assign to
the Reliance Team Assistant Director, another AD or the Director.
13. Once the Designated Reviewer has submitted the review, the site will appear in the “Post-Review”
state.
14. Click “Finalize Documents” and finalize the site’s consent form. Click OK.
15. Prepare the site’s approval letter using the Site Approval template from the drop down.
16. Click on the draft letter.
17. Add post nominals if necessary and edit the letter as needed.
18. Upload the revised letter.
19. Send the letter.
20. The site will now appear in the Active state.
21. Upload the approval documents for each site in IREx.
22. Repeat the process for each site that is to be added to the study.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
04/06/2022
Revised to reflect new process (no need for external team member list)
04/22/2022
Archived log of updates over 1 year old
02/24/2023
Minor clarifications such as studies being assigned to reliance team for
processing

Table of Contents
Page 62 of 273
SOP Title:
Processing Modifications When Emory is the Single IRB of Record (sIRB) for a Multi-
Site Study
SOP Category:
Collaborative Research / Central IRBs
Established:
01/20/2021
Last Revision:
08/15/2024
PURPOSE
This SOP outlines the steps to process modifications when Emory IRB is serving as the sIRB for external
sites enrolling participants.
SCOPE
This SOP applies to multi-site studies for which Emory has agreed to serve as the sIRB.
Process:
1. For modifications to add or remove Emory study personnel, follow the SOP “Adding/Removing Study
Personnel.”
2. For modifications to add or remove external site study personnel, the external study team is
responsible for ensuring study team members maintain current CITI and credentials in accordance
with their institution’s policies and procedures. Emory IRB only reviews the PI for each external site.
If the PI for the site changes, the relying IRB must complete a new local context review form or
provide confirmation via email that the new PI has met all local training requirements and can be
added as PI. If this is provided via email, the email must be uploaded in the modification.
3. For modifications indicating increased risk, follow the SOP titled “Modifications: Mods indicating
increased risk.”
4. For modifications that require new ancillary reviews such as a new COI, change to radiation
procedures, infectious agents, etc., a new local context review form must be obtained from each
relying IRB and the completed forms must be uploaded in the modification prior to approval. The
local context review form is titled “Emory sIRB Relying Site LCR Form” and is located in the folder
H:\External IRB Relationships 6.3.2020\06. Forms and Checklists.
5. If the study is an S-I study, trigger ancillary review by Margaret Huber if applicable (see S-I SOP for
guidance.)
If the Emory study team requests to add a new pSite, contact the reliance team lead to discuss. If it is an
S-I study, trigger ancillary review for Margaret Huber to approve new site.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/30/21
Clarity in purpose
04/06/2022
Added language pertaining to S-I studies
01/05/2023
Minor edits for clarity
08/15/2024
Remove guidance to reassign to an S-I specialist

Table of Contents
Page 63 of 273
SOP Title: Processing CRs When Emory is the Single IRB of Record (sIRB) for a Multi-Site Study
SOP Category:
Collaborative Research / Central IRBs
Established:
01/20/2021
Last Revision:
08/15/2024
PURPOSE
This SOP outlines the steps to process CRs when Emory IRB is serving as the sIRB for external sites.
SCOPE
This SOP applies to multi-site studies for which Emory has agreed to serve as the sIRB.
Process:
1. The reliance analyst will follow the SOP titled “Continuing Review Processing-Preliminary Analysis
through Approval” to process the Emory site CR.
2. The Emory study coordinator will distribute the “pSite Continuing Review Form” to each pSite to
complete and return to the study coordinator.
3. The Emory study coordinator will enter each site’s information from their completed CR form into
the spreadsheet titled “Continuing Review Report for sIRB Study.” Once the spreadsheet is
complete, the study coordinator will upload the spreadsheet in the supporting documents section of
the Emory site’s CR in SaaS. The forms and spreadsheet are in the folder H:\External IRB
Relationships 6.3.2020\06. Forms and Checklists.
4. While in the CR workspace, click “Sites”, select the site, then “Report Continuing Review Data” and
enter the information provided by the study team in the pSite Continuing Review Form. Upload the
completed form in the Supporting Documents section. The sites will now appear as shown below.
5. Mark yes for “Report Completed” for each site.
6. Once the study is reapproved, follow the steps outlined in the SOP “Continuing Review Processing-
Preliminary Analysis through Approval” to finalize documents, prepare letter and send letter.
7. From the CR workspace click “Sites” and then select the site.
8. Click Return to Post-Review and enter comment “Continuing Review Update.”
9. Click Submit Site Designated Review and enter the determination and dates from the main study CR
approval. Enter the same approval and effective dates unless the specific site was approved with
modifications and the modifications were approved at a different time.
10. Complete the rest of the Site Designated Review form and click OK.
11. Finalize Documents
12. Prepare the letter. There is only one letter template available for sites which is the initial approval.
Modify the letter template to mirror the Emory site CR approval letter but with the site information.

Table of Contents
Page 64 of 273
(Change title of the letter, add post nominals to PI, use the same approval language as the Emory
site, etc.)
13. Send the letter.
14. The site should now be in an Active state with the same approval and expiration dates as the Emory
site.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/30/2021
Added steps for re-approval of pSites
04/06/2022
Added guidance for S-I studies
02/24/2023
Minor edits for clarity
08/15/2024
Removed guidance for S-I review
IRB MEMBER MANAGEMENT

Table of Contents
Page 65 of 273
SOP Title: IRB Member Onboarding
SOP Category:
Meeting and Member Support
Established:
02/25/2021
Last Revision:
6/9/2021
PURPOSE
The purpose of this document is to detail activities from the time an individual becomes a member
candidate until appointment and first official IRB meeting.
SCOPE
The SOP applies to IRB staff working with the IRB Member Onboarding process.
DEFINITIONS
Member Candidate: An individual who has submitted a completed application, a signed IRB
Confidentiality Agreement and a copy of his/her CV or resume to apply for IRB membership.
Appointment Date: Date on the appointment letter returned with the signature of the Emory
Institutional Officer (I.O.)
PROCEDURES
IRB Director/Assoc/Asst Director:
1. Conduct an informal 3-5 minute phone call. Confirm the individual’s interest in membership,
prior IRB experience if any, expertise and availability. OR send the following recruitment email
to potential candidate if no phone number is available:
a. H:\General\CMTE\2. Members and Roster Docs\Recruitment - New
Members\Application Materials for Membership\2. Email Templates\Community_
Member_UA-NS-Recruitment Email Text.docx
2. Once interest and eligibility is confirmed in consultation with the Director, send the relevant
Emory IRB Membership email outlining the appointment process (either the one for Emory-
affiliated members, or for Community members):
a. Template: H:\General\CMTE\2. Members and Roster Docs\Recruitment - New
Members\Application Materials for Membership\2. Email Templates
Attach copies of the following to the email:
• IRB Member Application Fillable Form (latest version date)
• IRB Confidentiality Agreement Form
• Instructions for completion of the CITI Member Module “What Every New IRB Member
Needs To Know”* Or CITI IRB Community
Member_New_Account_&_module_training_document
• IRB New Member Pre-requisites document OR IRB New Member Pre-requisites-
community members document
• New Member Orientation-(latest version)
• **Community Members only – Supplier_Individual_Information Form & Guidance for
filling in vendor form for community members

Table of Contents
Page 66 of 273
The above documents are attached to the email template or can be found here:
H:\General\CMTE\2. Members and Roster Docs\Recruitment - New Members\Application
Materials for Membership\3. Forms and Documents
3. Update a copy of the New Member Onboarding Checklist with date materials were returned or
trainings completed.
4. Create folder for the member in the H: drive (H:\irb_shared\General\CMTE\2. Members and
Roster Docs\Individual Member Files\01. Appt-Resig and CVs - by member) and store signed
appointment letter, CV, and completed application, and copy of New Member Onboarding
Checklist. You may wish to copy a shortcut to the checklist onto your Desktop until onboarding
is complete.
5. Ask future Pod leader to set up a date and time for the candidate’s orientation and training of
the IRB/eIRB system at the IRB offices or over Zoom. Email time, place and logistics to the
candidate.
Relevant Pod Leader (loop in Director re: all steps, so that Roster can be updated promptly):
6. Update the New Member Onboarding Checklist with the scheduled training date.
7. Send a reminder email to the candidate before the scheduled training with review of the
logistics.
8. Conduct the New Member orientation training using the New Member Power Point.
H:\General\CMTE\2. Members and Roster Docs\Recruitment - New Members\Application
Materials for Membership\4. Training Materials
Key Points to include and/or emphasize:
• Emory IRB Committee (A/B panels and Team C)
• Meeting time and place
• Meeting structure/presentations
• Quorum
• Member attendance once monthly and completion of assigned reviews in the eIRB
system
• Review of a NEW study, CR and AM in the system
• The schedule for Reviewer Assignments Completion
• Member commitment to serve 2 years with option to renew for a third year
• Role of Meeting Pod
• Role of IRB Staff Analyst/Study Owner
• Decide the panel assignment with the candidate.
• Set up a meeting observation date.
• Meet IRB Director if available.
• Show conference room 5C when walking candidate out to the elevators
9. Update the Director and the New Member Onboarding Checklist with date of training
completion and scheduled date for meeting observation.
10. Email candidate on what to expect after the observation. Confirm panel assignment.
11. Update the New Member Onboarding Checklist with observation completion date.
12. Draft appointment letter (template). Request review by the Director.
13. After Director review, email the appt letter to the office of the I.O. with a copy of the
candidate’s CV/Resume.
14. Update the New Member Onboarding Checklist with the date sent to the I.O.’s office.

Table of Contents
Page 67 of 273
15. Update the New Member Onboarding Checklist when the signed letter is returned to the IRB
with the official appointment date (date on letter).
16. Email official welcome to the new member with appointment letter attachment and instructions
for acknowledgment.
Copy the following persons on the welcome email:
IRB Director
IRB Co-Chairs
Meeting Chair
Team Lead
All Pod members
17. Confirm all documents (including fully signed appointment letter) have been stored in the
member’s folder on H: drive
18. If the new member is unaffiliated, request a sponsored account for her/him through IT Service
Now.
19. Submit IT request for the user to receive the appropriate Role in eIRB
20. Update eIRB system to list new member on the appropriate panel. (Anyone in Pod can do this)
IRB Director:
21. Update member roster per instructions on Roster’s first sheet
22. Add new member to the Outlook member group email listing (new).
a. Find list here: H:\General\CMTE\2. Members and Roster Docs\All Member Email List -
update as needed.
b. Open file – Click on “Add Members” and “Select Members: Offline Global Address List.”
Search by last name.
c. Highlight the member you are adding, then click the “Members” button at the bottom
of window. The members name will be added to field, then click “OK.”
d. Save & close mailing list.
e. Open new message in your Outlook and copy the updated “IRB All Member Mailing
Group – Updated ______(date)” to the email.
f. Send email to the IRB Staff.
23. Confer with appropriate meeting pod on date of first official meeting and how the candidate
faired.
24. Update member roster with first meeting date and any missing information to close out former
candidate tracking.
25. Announce new member at next IRB staff meeting.
PROCESS FLOW

Table of Contents
Page 68 of 273
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
6/9/2021
Removing an old statement
Member
candidate
returns
application,
confidentiality
document, and
CV/Resume.
IRB/eIRB
Member
Overview and
Meeting
Observation
Appointment
and Set-up
Documentation
and First
Meeting

Table of Contents
Page 69 of 273
QA AND EDUCATION
SOP Title:
Acknowledgments & Noncompliance Determinations Made by Senior Team Q Staff
SOP Category:
QA and Education
Established:
10/19/2011
Last Revision:
6/9/2021
PURPOSE
The purpose of this document is to specify in which circumstances a designated senior Team Q staff may
acknowledge reportable new information submission submitted to the Emory IRB.
SCOPE OF SOP
The SOP applies to the reportable new information submissions submitted for studies reviewed by the
Emory IRB.
RESPONSIBILITIES
• Senior team Q staff: Reviews and triages reportable new information submissions submitted to
the Emory IRB. Under this SOP, the staff will also acknowledge certain type of events as
explained below. “Senior” is defined as Associate or Assistant Director, or other Q staff
designated by the Director as having sufficient experience and expertise to be considered
“Senior.”
• Non-senior team Q staff: May administratively process acknowledgments on behalf of senior
team Q staff, but would not make the determination themselves.
PROCEDURE
The designated senior Team Q staff is allowed to acknowledge the events as detailed in this SOP. The
designated senior Team Q staff may, at his/her discretion, send any of these events to an IRB vice-chair
even if noted in this SOP to get confirmation of appropriate review. The designated senior Team Q staff
may acknowledge the following events:
o Study staff not added to IRB submission, as long as staff was proper research (CITI and, if applies,
Emory Clinical Research training) and protocol training before starting study activities
o ICF/HIPAA documentation issues where:
The form used was expired but it was the correct version approved by the IRB
The subject signing the consent forgot to time and date. A note to file is required to clarify this
matter.
The study team member forgot to time and date the signature. A note to file is required to
clarify this matter.
Discrepancies in signature/date/time by subject or study team unless there is a pattern of
noncompliance.
ICF missing fields in the consent form that do not involve options made by the subject or
signature, e.g. initials in pages.
o Lapses in approval for FDA trials where research activities did not take place during the lapse. Other

Table of Contents
Page 70 of 273
studies do not need the submission of a RE.
o Errors in reporting enrollment numbers at continuing review (whichever is greater):
Gap difference in enrolment number is within 10, no matter sample size
No greater than 20 % of total enrolled.
o Over-enrollment in a NMTMR study, when subjects have undergone study procedures (not only
signing consent), as long as it is the first occurrence for a study, if HIPAA does not apply.
o Any event caused by the subject’s (not the study team’s) lack of adherence to the protocol that, in
the opinion of the principal investigator, does not affect the subject’s safety, rights and welfare or
willingness to continue with study participation, and is not an unanticipated problem.
o Adverse event (that is not an internal death) that is considered unanticipated by the principal
investigator but for which the causal relationship to study participation is unknown, and no more
information is or will be available. Such events will be acknowledged with directions to submit a
new reportable new information submission if the cause of the event is determined as related at a
later date.
o Adverse event reported to the IRB per sponsor requirements, that the principal
investigator considers anticipated or unrelated to study participation. For VA studies, this
will need to be acknowledged by a VA reviewer.
o Protocol deviation which the principal investigator considers not substantive and not affecting
subjects’ rights, welfare, safety, or willingness to continue with study participation, and not
affecting integrity of the research data, reported per sponsor requirements.
o DSMB letter indicating that study can continue per the protocol without change, submitted per
sponsor requirement only.
o Protocol deviation that may minimally affect the integrity of research data, but does not affect
subjects’ rights, welfare, or safety and which does not represent a pattern of noncompliance.
Team Q staff should review prior reports and consult the vice-chairs when in doubt.
Examples of such deviations are:
Visit occurred out of window.
Test done for research (not for safety) purposes which was not drawn or was drawn out of
window, which does not reflect a pattern of noncompliance.
Missing data caused by subjects’ noncompliance with protocol (specifically, missed data when not
completing surveys not used for diagnosis or treatment) or missing data due to programming
errors that do not affect subjects’ safety.
Surveys or survey items completed in error, when the surveys or survey items’ completion does
not negatively affect subjects’ rights, welfare or safety. For example, the completion of a survey
asking for information that may upset the study participant should be sent to a vice-chair
reviewer.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
1/4/2016
Change in the procedure section
6/28/2016
Change in the procedure section
7/14/2017
Changes to procedure section
6/9/2021
Adding a new criterion for acknowledgment

Table of Contents
Page 71 of 273
SOP Title:
IRB Noncompliance
SOP Category:
QA and Education
Established:
3/24/2015
Last Revision:
08/15/2024
PURPOSE
The purpose of this SOP is to describe the process of reviewing situations in which the IRB, either
through the actions of the IRB Committee or its administrative office, may not have followed applicable
regulations or its internal policies and procedures; this policy does not apply to instances of non-
compliance by investigators or research team members. (NC). The Emory AVP of the HRPP, the Emory
IO, and the ORA Office of Research Compliance and Regulatory Affairs (RCRA) are tasked with making
sure the Emory IRB complies with the IRB policies and procedures and applicable federal regulations.
SCOPE
The SOP applies to all actions and determinations taken by the IRB.
DEFINITIONS
• IRB Noncompliance (NC): a failure of the IRB to follow IRB Policies and Procedures, IRB Internal
Standard Operations Procedures or federal regulations during the review and oversight of study
submissions.
• Minor IRB NC: IRB NC that does not significantly impact the rights, welfare, safety of participants, or
the integrity of the research data.
• More than Minor IRB NC: IRB NC, or identification of a repeated pattern of Minor IRB NC, that could
significantly impact the rights, welfare, safety of participants, or errors that cause a disruption for
the study team that would take significant steps to resolve (for example, a large number of subjects
will need to be reconsented)
• Findings: issues discovered during an IRB internal QA/QI process that may indicate an error during a
study submission’s review and approval by the IRB. A finding would not rise to the level of minor or
more than minor noncompliance if it does not affect the rights, welfare, safety of participants, or
the integrity of the research data.
RESPONSIBILITIES
• AVP for (RCRA) and Director of the Office of Research Integrity and Compliance (ORIC) – after the
IRB identified trends, reviews information with the INS before routing to the IO.
• AVP of the HRPP – reviews IRB NC and makes a determination as to whether an event is minor or
more than minor.
• IRB Q Team: Compiles information to be sent to the AVP of the HRPP or IRB NC Subcommittee (INS)
or the AVP for RCRA and Director of ORIC as applicable.
• IRB NC Subcommittee (INS): reviews issues coming from the AVP of the HRPP (through a Team Q
representative). The IRB INS is composed of the AVP of the HRPP, IRB Co-Chairs, and the AD who
supervises the IRB analyst for the affected submission (if applicable). INS’s role is to determine the

Table of Contents
Page 72 of 273
CAPA is adequate for any IRB Noncompliance that is more than minor. They also determine whether
a trend is more than minor IRB NC.
• AVP of the HRPP and Associate or Assistant Directors (ADs) – Utilizes IRB NC data to determine if
there is a trend related to findings from routine record reviews, or other issues identified by IRB
staff, researchers, or other members of the HRPP.
• IO: reviews more than minor IRB noncompliance to assess reporting requirements and adequacy of
Corrective and Preventive Action (CAPA) plan.
PROCEDURE
1. IRB noncompliance issues may be identified during routine internal review or during incident-based
reviews by IRB staff, researchers, or other members of the HRPP. An AD or Q team member will log
all potential IRB NC in the IRB NC spreadsheet.
2. All potential IRB NC (that hasn’t previously been determined to be no more than minor) should be
elegantly summarized and sent via email to the AVP for the HRPP. Include links in eIRB if relevant.
The AVP for the HRPP will determine if findings constitute IRB NC and will determine which are
minor and which are more than minor.
3. If the matter constitutes no more than minor (NMM)IRB NC, a CAPA will be instituted as applicable.
4. If the matter constitutes more than minor IRB NC, the AVP will email the Team Q representatives to
route the matter to INS and the IO.
o A root cause analysis and a CAPA plan must be drawn up for each matter that is potentially
more than minor (MM) IRB NC. The Team Q representative should work with the parties
involved to document this before sending to INS and the IO.
Cumulative Report Process for minor IRB NC
1. All minor IRB NC will be logged by an AD or Q team member in this spreadsheet
located at
OneDrive/IRB-Staff/Documents/H drive/General.
2. Team Q will review this spreadsheet routinely to identify trends.
o If a trend is identified, a Team Q representative will provide a summary of the trend, the trend’s
impacts, and CAPA to INS via email. Reminder to copy: the AVP for (RCRA) and Director of the
Office of Research Integrity and Compliance (ORIC)
The INS will make the determination of whether the trend is no more than minor or
more than minor noncompliance and will determine whether the CAPA is adequate.
If more than minor, route to IO per SOP steps.
If no more than minor, close out per SOP steps.
o If no trends are identified, document the absence of trends in the spreadsheet.
INS Review
The below process will be followed for identified trends during the review of the spreadsheet
information.
o A summary with relevant details will be provided including trend information
o To close the case, the AVP for RCRA (or designee) must vote for no more than minor IRB NC, and
at least two members of INS must agree.

Table of Contents
Page 73 of 273
o The event will be updated in the spreadsheet and the case will be considered closed when the
CAPA plan is completed, if applicable.
o After receiving all required responses to close the email communication, the email thread
should be saved under H:\General\QA Working Files\NC UP Complaints\IRB NC\Emails sent to
INS with cumulative reports. The email thread should have the date of when this information
was sent to the INS
3. If the INS determines that the case constitute more than minor IRB noncompliance, follow the
process under “IO Review”.
IO Review
1. If the responses indicate that the cumulative report or case information needs to be sent to the IO,
forward the INS email thread, and any other related information to the IO for their review. Copy the
AVP of the HRPP, AVP for RCRA and ORIC Director in this communication. Specify in the email that
you have sent this information to the INS and that you need them to decide if the noncompliance
should be reported to the federal oversight agencies.
2. The IO will review the information to assess reporting requirements and adequacy of the CAPA plan.
a) If the IO determines the case requires reporting, the Team Q representative will work with the
AVP of the HRPP and IO in creating a letter to the oversight federal agencies, and AAHRPP, as
applicable. Update the spreadsheet
.
b) If the case does not require reporting, update the spreadsheet and save the communication
from the IO in this folder: H:\General\QA Working Files\NC UP Complaints\IRB NC\IO review.
PROCESS FLOW
Each individual incident of IRB NC:
Ongoing Trending
AVP of the HRPP
reviews events of
potential IRB NC
and makes
determination
Minor IRB NC
events are logged
in spreadsheet,
and more than
minor IRB NC
events are sent to
INS and the IO by
Team Q
representative.
IO reviews for
reporting needs
and adequacy of
CAPA plan. INS
reviews adequacy
of CAPA plan.
Outcomes
documented on
the spreadsheet
and H drive as
applicable. Team Q
representative
assists with
reporting if
needed.

Table of Contents
Page 74 of 273
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
07/06/2023
Clarity on spreadsheet and archived changes over a year old
8/15/2024
Clarity of processing trends
Team Q reviews IRB NC
in the spreadsheet for
trends
If a trend is identified, a
Team Q representative
routes to INS for a
determination. If no
trends are identifed,
Team Q documents
absence of trends in
spreadsheet.
INS votes on whether
trend is NMM or MM
IRB NC
Trends that are MM IRB
NC are sent to IO for
review. IO reviews
adequacy of CAPA and
determines if other
reporting is needed.
Outcomes documented
on the spreadsheet and
H drive as applicable.
Team Q representative
assists with reporting if
needed.

Table of Contents
Page 75 of 273
SOP Title:
IRB Team Q CAPA Follow Up
SOP Category:
QA and Education
Established:
2/14/2012
Last Revision:
5/24/2021
PURPOSE
The purpose of this document is to describe how the IRB Team Q will follow up on the IRB-approved
Corrective and Preventive Action (CAPA) plans presented during Committee Q meetings. In addition,
this SOP will cover the process of auditing CAPA plans for studies conducted at Emory and reviewed by
External IRBs.
SCOPE
CAPA plans from studies reviewed by an External IRB or during Emory IRB Committee Q meetings for
possible serious (SNC) and/or continuing non-compliance (CNC), and unanticipated problems (UP)
resulting from study staff oversight or error.
DEFINITIONS
• Corrective and Preventive Action Plan – a plan developed by an investigator, with or without the
assistance and guidance of the HRPP, following a root cause analysis into an instance of
noncompliance or other problems in the conduct of human subjects research. The CAPA must
include measures designed to correct the immediate problem and prevent its recurrence or the
recurrence of a similar type of problem. CAPA plans are reviewed and may be modified by the IRB
before being approved. Investigators are responsible for implementing CAPAs in a timely manner.
• Committee Q – Full Board Committee that reviews cases of possible serious non-compliance,
unanticipated problems, and conflict of interest issues for ongoing studies. The COI committee
prepares management plans for review by the IRB who has authority for final approval.
• Team Q: IRB staff team specializes in Education and QA efforts including non-compliance,
unanticipated problems and protocol deviation review, analysis, data-gathering and presentation.
• CoRe team: A designated group of the IRB Chair, Director, and qualified IRB staff to investigate cases
of alleged non-compliance and UPs. Their findings are documented as part of the IRB record. All
cases of non-compliance, UPs, and suspensions and terminations will be investigated and followed
by the CoRe team. Additional investigations by other units or individuals may proceed concurrently
or in sequence with those of the CoRe team.
RESPONSIBILITIES
• Committee Q –reviews, request changes in, approve or disapprove CAPA plans, and follows-up
reports at Committee Q meetings.
• Team Q – Team Q designated member will compile CAPA updates. The follow-up report will be
presented at the Committee Q meeting every month. Every Team Q case manager will email the
designated person CAPA updates. The Team Q designated member will add the information to the
follow-up report for the next available Committee Q meeting.
• Study Staff: Responsible to complete CAPA plan in the time allowed by the Committee Q members.
This period is normally 30 calendar days from the date of the meeting unless specified otherwise.
• External IRB: Provides information about determinations of SNC, CNC, and UPs for studies
conducted at Emory University/Healthcare.
PROCEDURE

Table of Contents
Page 76 of 273
Steps for Studies Reviewed by CMTE Q
• After Committee Q reviews and approves the CAPA plan for a specific case, the CAPA will be added
by answering yes to question 2 (is further action required) under “Submit RNI Committee Review”.
The person adding the notes will assign the RNI Action to the appropriate party and will click ok. The
RNI Action Plan will be included in the determination letter sent to the principal investigator
• After the meeting and the RNI will enter “Action Required” state.
• Every responsible party will follow up with the study team about the CAPA plan completion and the
deadline.
• When the CAPA plan (RNI Action Plan) is completed, the responsible party will submit an Action
Response in eIRB. The case manager will review the action response. The case manager will
complete the Required Actions Reviewed activity and will mark the action completed as required or
not.
• If the action is completed, the case manager will prepare and send an RNI Action Complete letter to
move the RNI to “Review Complete” status.
• If the CAPA plan has not been completed in the allowed period, the case manager will notify the
study team that non-completion by this deadline is deemed non-compliance and will request an
explanation of the delay to be submitted along with a notice of completion. This new NC will be
reviewed by CoRe. The CoRe team will be notified about the delay for any additional determination.
The Team Q designated member will create a report of incomplete CAPAs for the Full Board after
CoRe review if this is considered reportable to FB.
Extension for CAPA Plan
The study team may request a deadline extension. This extension may be reviewed and granted or
denied at the CoRe team discretion.
Steps for Studies Reviewed by an External IRB
• The reliance team will inform Team Q of an external IRB determination of SNC, CNC or UP.
• Team Q may review the CAPA plan and schedule an audit to check the progress with the
implementation of the CAPA
o The audit can be conducted in person or via email correspondence, depending on the case
and/or CAPA plan. For example, if the CAPA plan requires the creation of a document, Team Q
can request a copy and examples of when it was used via email.
REFERENCES
• IRB QA Plan 12.15. 08
• 45CFR 46.
• 21CFR 820.
• IRB policies and procedures
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/11/2020
Updates to align with new system
3/4/2020
Additional changes for clarification
5/24/2021
Added procedure for studies reviewed by External IRBs

Table of Contents
Page 77 of 273
SOP Title:
CMTE Q Meeting Facilitation
SOP Category:
QA and Education
Established:
6/11/2013
Last Revision:
12/11/2020
PURPOSE
Outline the necessary steps of facilitating a Committee Q meeting; before, during, and after the
meeting.
SCOPE
Applies to Team Q and administrative staff who help conduct Committee Q meetings.
PROCEDURE
Q RPA
• creates a folder in H drive
• sends invitation emails
• keeps track of quorum using CMTE Q attendance Workbook
• preps for the meeting using CMTE Q Recap and Prep workbook
• works with Sr. RPA and using RAS assigns reviews in eIRB
• sends agenda email
• sends email reminders to IRB staff
• sets up for meeting
• drives during the meeting (completes omnibus forms as needed)
• updates attendance in attendance tracking sheet and emails Carol Corkran for
Payment
• sends minutes to members
Q Sr. RPA
• collaborates with RPA on reviewer assignments
• takes notes during the meeting (can be in OneNote or a word document)
•
polishes minutes before sending to the AD or Director for review
• leads the pre-meeting and post-meeting Pod meetings
• takes backup notes during the meeting
• advises pod members as needed
Directors
• takes backup notes during the meeting
•
reviews final version of minutes for QA/QI purposes
Before the Meeting
1. The Q RPA creates a meeting folder in H:\IRB\General\QA Working Files\CMTE Q\YYYY. The folder
title should be MM-DD-YYYY.
2. Create a meeting agenda and save it in the meeting folder. Individual items can be added as they
are identified. A copy of the worksheets, plus any additional materials should be saved in PDF
format under the “Meeting Materials” folder. The “RNI Full Board Document” should be saved
under “FB Forms”. The meeting agenda is populated to the meeting space by clicking on prepare
agenda, and then uploading the agenda under question 2 by clicking on the ellipsis and selecting
“upload revision”.

Table of Contents
Page 78 of 273
3. Each meeting folder should include subfolders for PI letters, Fed Letters, Investigator slot
spreadsheets, Meeting Notes, Meeting Materials, and FB forms. The Recap Spreadsheet is located
under the YYYY folder.
4. The Q RPA uses the CMTE Q Recap and Prep Workbook to ensure documentation from prior
meeting is complete, including CAPA and federal notifications.
5. During the first week of the month, the Q RPA sends an email invitation to all members to ensure
quorum and get an accurate count of attendance. Each committee may have a maximum of five (5)
physician scientists, a maximum of four (4) non-physician scientists and must have one (1) non-
scientist and one (1) unaffiliated community member present(the non-scientist and unaffiliated
scientist may be the same person). It is preferable to have a VA member present if VA cases are
reviewed, but it is not required. Any identified conflicts of interest should be taken into account
when ensuring quorum. To assign reviewers:
5.1. Click on “Assign Reviewers” under the meeting space
5.2. Click on “update” to each RNI and add the reviewers
5.3. If there are other agenda items that need to be reviewed, assign a reviewer if applicable
5.4. Say yes to question 3 to notify members of their assignments, if ready. If not ready, say no,
and send when ready.
6. The agenda should be finalized and materials sent to all attending members no later than the Friday
before the meeting, if possible using the meeting space. The VA Compliance Officer and Director of
Research/IRB Director at CHOA should receive an invitation to the meeting, and a copy of the
VA/CHOA materials to be reviewed, if applicable. Guest arrivals should be noted on the agenda (for
example, Dr. Jones and Ms. Smith will be present at 4:15 pm). The case manager of each study is
responsible for inviting guests, providing directions, and noting arrivals on the agenda.
7. If any items are added or removed after the agenda and materials are sent, the members should be
notified via email and through the system as soon as possible and provided any additional materials
that are needed. New materials pertaining to a specific case may to all members. The case manager
should notify the meeting facilitator when these changes have occurred.
8. If there will be call-ins to the meeting, the caller should be given the appropriate phone number.
The meeting facilitator is responsible for setting up the teleconference equipment or teleconference
electronic meeting. If the meeting takes place online, the waiting room must be enabled to allow
guests in only at the appropriate time.
Day of the Meeting
1. The administrative staff will set up drinks and snacks about 30 minutes before the meeting.
2. The Q RPA will place a sign near the elevator to indicate PIs attending the meeting to wait in room
5A.
3. The Q RPA will make all meeting materials available (as a viewer) to attending committee members
on Emory Box.
4. The Q RPA will make available name cards and copy of the agenda for all committee members and
staff at the meeting.
5. The Q RPA will ensure that laptops are set up for members and staff.
6. If necessary, Q RPA will set up the speakerphone.
After the Meeting

Table of Contents
Page 79 of 273
1. IRB staff should work together to clean up after meeting, including putting away IT equipment and
locking the storage closet.
2. The laptop cart and other IRB equipment should be taken to the IT area for storage.
3. The day after the meeting, IRB staff will meet to recap the meeting and strategize plans for sending
determination letters. Each case manager is responsible for creating his/her own letter and
ensuring it is sent to the PI. If specific follow up activities are required (site visit, Modification
required, etc.), the case manager is responsible for that follow up. CAPA plans will be reviewed.
4. Letters to federal oversight agencies should be drafted concurrently with the follow-up letters to the
PIs. Send draft letters (FDA/OHRP) to the IRB Director for review. Occasionally, the Institutional
Official will also be asked to review and comment. Letters should be sent to the feds within 1
month. If the matter is considered of great urgency, the letter should be sent earlier as possible.
The IRB co-Chair or vice-chair with medical expertise should sign any letters to the FDA; the IRB
Director may sign letters to OHRP. A copy of the email sent to FDA and OHRP should be kept in the
“Letter to Feds” folder under the meeting date folder.
5. Minutes should be completed and sent to the Associate or Assistant Director for review at least one
week prior to the next meeting. Follow the Minutes SOP
for details about this process.
6. Every Team Q member will be responsible for follow- up with the study PI regarding CAPA
completion. The meeting facilitator should remind the Q team to add outstanding CAPA items to
the CAPA report worksheet in the meeting materials folder.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
11/3/2020
Clarified that only attending members should receive meeting information
12/11/2020
Added additional actions to be taken by the Q RPA; corrected steps for virtual meetings
10/14/2021
Replaced Julie with Carol regarding notification for community member payment and archived log of
updates over 1 year old

Table of Contents
Page 80 of 273
SOP Title: Communication of Report of Internal Study Subject Death
SOP Category:
Administrative
Established:
7/17/12
Last Revision:
1/21/2022
PURPOSE
The death of a subject who is an Emory Healthcare (EHC) Patient or University Employee or Student
while in a study raises major institutional concerns if the death is deemed unanticipated, related to
study participation, and potentially increasing the risk for participant s or others. This SOP is intended to
guide the IRB in communicating with EHC and University Officials.
SCOPE
Deaths considered unanticipated problems in Emory study participants
PROCEDURES
1. Assess whether we have enough information to notify others who need to know. In most cases, we
will need more information or clarification; in such case, the IRB Director, Education & QA Lead, or
Co-Chair should communicate directly with the PI, without cc’ing others who will want to know
about it once we have all the relevant information. The reason for this is to avoid premature
questions flooding in.
2. Recommend to the PI to let his clinical chair and any relevant research coordinators and study staff
know about the event and any immediate implications for other study subjects.
3. Ask the PI whether they know anything about whether the subject’s family is talking about bringing
suit against Emory or has other serious concerns about the study’s relatedness to the death. This
information should be included in the notification to Risk Management and OGC.
4. Once we have enough information to say whether the PI thinks it is related (probably, possibly, etc.)
or not, and whether the PI thinks it is anticipated or unanticipated, and whether he thinks the
protocol/ICF should be changed and subjects notified, the IRB Director or Education & QA Lead
should do the following:
a. Sent to CoRe for review.
b. If CoRe considers the event to be a possible unanticipated problem, the following people should
be alerted via email, (including the date when the full board will review the event):
i. Robert Nobles, VP for Research Administration
ii. Kimberly Eck, AVP for Research Compliance and Regulatory Affairs and RIO
iii. Maria Davila, Director for Research Compliance and Regulatory Affairs and RIO
iv. Margaret Huber, IRB and FDA Compliance Manager
v. David Stephens, VP for Research
vi. Jeffrey Lennox, Associate Dean for Clinical Research
vii. If Winship: Ramalingam, Waller, El-Rayes, Rodger, and PI
viii. Katie Lewis (OGC)
ix. Cheryl Ritchie (Risk Management)
x. Anne Adams and Stephanie deRijke (CTAC)

Table of Contents
Page 81 of 273
xi. Laura Deane (ORisk Management)
xii. William Bornstein (Emory Healthcare Chief Quality Officer)
xiii. Susan Rogers (IDS) – if it is a drug trial (Non-VA)
xiv. IRB analyst owner of the study
xv. Chair of IRB meeting that will be reviewing the possible UP
xvi. Anyone else it makes sense to alert (check with IRB Director first)
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/25/2021
Updates to reporting lines
5/24/2021
Updates to reporting lines
1/21/2022
Updates to reporting lines and archiving log of changes over 1 year old

Table of Contents
Page 82 of 273
SOP Title:
Review of Single Use, Expanded Access of Unapproved Drugs or Devices
SOP Category:
QA and Education
Established:
7/27/12
Last Revision:
12/11/2020
BACKGROUND
The FDA allows the use of an unapproved medical drug or device both for research and under what is
known as expanded access or compassionate uses. This allows for the use of drugs or devices for
treatment purposes outside a research protocol.
PURPOSE
The purpose of this document is to describe the review process of a new study for the use of an
unapproved (or not approved for a specific indication) drug or device for Compassionate or Emergency
use, from submission to approval.
SCOPE
Compassionate/Emergency use of an unapproved drug or device in an Emory study participant or
patient, done by an Emory affiliated physician. If the use is submitted from a doctor without an Emory
affiliation, even if treating a patient in an Emory facility, it would be reviewed by WIRB or another
external IRB. Expanded access uses at CHOA should be routed to the CHOA IRB.
DEFINITIONS
• Sponsor: A person who takes responsibility for and initiates a clinical investigation. The sponsor
may be an individual or pharmaceutical company, governmental agency, academic institution,
private organization, or other organization. The sponsor does not actually conduct the investigation
unless the sponsor is a sponsor-investigator
• Sponsor-Investigator (S-I): means an individual who both initiates and conducts an investigation,
and under whose immediate direction the investigational drug is administered or dispensed. The
term does not include any person other than an individual. The requirements applicable to a
sponsor-investigator under this part include both those applicable to an investigator and a sponsor
RESPONSIBILITIES
• IRB Team member: facilitates the submission of compassionate and emergency use request,
verifying that the information provided is complete.
• IRB Co-Chair: reviews compassionate or emergency use requests before full-board review.
• Investigator: makes sure that the information submitted is complete and accurate, according to the
information submitted to the FDA and/or the sponsor.
• Sponsor: submits an IND/IDE supplement to the FDA before use to obtain permission (for
compassionate use only), and an IDE supplement after use as a follow-up report.
PROCEDURE
NOTE: Make sure to use our letter templates for IRB Chair concurrence, IRB acknowledgment of use or
IRB approvals for these compassionate uses. The Team Q member should review the submitted
information, and assist investigators when possible, directing them to the Emory policies and
procedures, FDA guidance or to the Research Integrity and Compliance for the submission of the
IND/IDE supplement (in case of an Emory Sponsor-Investigator).

Table of Contents
Page 83 of 273
Emergency use of a device: does not require IRB or FDA approval before its use but the study team
needs to contact the FDA to inform them of the use. In addition, the study team should submit an
application to the IRB for acknowledgment of this use. This will be reviewed at Full Board for
acknowledgment. The application should contain information about all the protection measures used
before use and the following:
• A description of the circumstances that required the use.
• IDE protocol with a description of the device and name of IDE holder.
• Copy of uninvolved physician’s assessment of use
• Copy of authorization from IDE holder if applicable
• Copy of consent document used for expanded access use (template use OK)
Before using the device, the physician should take as many of the following patient protection measures
as possible, and provide the following information in the submission:
• Obtain a written independent assessment of the use of the device by an uninvolved physician
• Obtain documented informed consent from the patient or his/her Authorized Legal Representative
• Obtain documented authorization from the holder of the IDE for the Investigational Medical Device,
if an IDE exists.
• Notify the Emory IRB by contacting the IRB Chair or his/her designee, and provide the Emory IRB
with a written description of the circumstances necessitating the use of the device, along with
copies of the uninvolved physician’s assessment, informed consent and the IDE’s holder’s
authorization.
• Notify any other institutional officials who require notice under institutional policies.
• If the patient did not consent before the use of the device, the physician should document this
matter as described under the Emory IRB P&P, entitled Waiver or Alteration of Informed Consent for
Research.
After the Emergency use submission has been initially reviewed, the Team Q member will contact the
study team in case of any required changes, while alerting the IRB Chair via email about the Emergency
use. Once ready for review by the full board, the Team Q member will place the study in the next
available meeting agenda, notifying the meeting facilitator and meeting Vice-chair. The Team Q
member will assign the review of the Emergency Use to the meeting Vice-Chair and the IRB chair.
After the Emergency use is approved by the IRB FB, the IRB approval letter should be created and it
should include the following:
• The patient should be followed-up for any unanticipated adverse device effects
• The treating physician should inform the IDE sponsor of the patient status after receiving the device,
so they can comply with their IDE reporting obligations
• If an IDE does not exist, the treating physician should provide the FDA with a written summary of the
conditions constituent the emergency, patient protection measures taken and patient results
• Evaluate the likelihood of similar use of the device occurring again at Emory, and if such future is
likely, begin steps to obtain a new IDE or a modification to an existing IDE to cover the device’s
future use and to obtain Emory IRB approval.
Compassionate use of a device: should be approved by the FDA before the use. The use will also need
IRB approval or, alternatively, IRB chair concurrence of use. If the IRB chair determines that the use

Table of Contents
Page 84 of 273
would benefit from expedited review, the use would be routed that way, even if IRB chair concurrence
was requested.
If the study team is seeking IRB Chair concurrence, the following information is required:
• A description of the circumstances necessitating the use.
• IDE protocol with a description of the device and name of IDE holder.
• Copy of uninvolved physician’s assessment of use.
• Copy of authorization from IDE holder.
• Copy of consent document for expanded access use (using our current template)
The Team Q member is required to do the following:
• Create a folder
with the information received from the treatment team. To document the use in
the system, the study team will create an RNI submission and will add all the documents required
for the concurrence.
• Contact the study team in case of any required changes, while alerting the IRB Chair about the
Compassionate use.
• Redact any identified information in the RNI submission and upload revised copies to the
submission.
• Send the concurrence to the study team after the IRB chair confirms it in the submitted RNI
The physician should not use the drug or device unless and until FDA approval of the Compassionate Use
and IRB concurrence (or Approval, in the case of a Compassionate Use for a group of patients) has been
obtained. If the FDA approves the Compassionate Use and the IRB concurs, then the use may occur.
If the study team is submitting to the IRB instead of asking for IRB chair concurrence, or if the IRB chair
decides the use should be reviewed via expedited review, the Team Q member will assign the study to
the IRB clinical chair or vice-chair and process as normal.
After the approval of the device use, the IRB approval letter should include the following
1
:
• The physician should develop a monitoring schedule for the patient and follow it in an effort to
detect any possible problems arising from the use of the device.
• The physician should prepare a follow-up report on the use of the device, including a summary of
patient outcome and a description of any problems encountered using the device. This report
should be provided to the IRB within 5 days of the use, as well as to the sponsor.
• The Sponsor should provide the FDA with a copy of the follow-up report as an IDE supplement.
• If the compassionate use has not been approved by the FDA, the letter should state that the device
cannot be used until FDA approval.
Emergency use of unapproved drug: This use does not need to be approved by the IRB although
requires authorization by the FDA. Authorization of the emergency use may be given by an FDA official
by telephone, provided the physician explains how the expanded access use will meet the requirements
and agrees to submit an expanded access application within 15 working days of FDA’s initial
authorization of the expanded access use. The physician may choose to use
FDA Form 3926 for the
1
https://www.fda.gov/medical-devices/investigational-device-exemption-ide/expanded-access-medical-devices

Table of Contents
Page 85 of 273
expanded access application. The IRB will require a full submission within 5 working days of the use.
This will be acknowledged by the Full Board. The submission should include:
• A copy of all information submitted to the FDA in connection with the Expanded Access use request.
• Informed consent form to be used or information demonstrating qualification for Emergency Use
exception from informed consent. See the P&P entitled: Waiver or Alteration of Informed consent
for Research, subsection entitled Emergency Medical Care Exception – Exception to the
Requirement to Obtain Informed Consent for the Use of a FDA-Regulated Item in Emergency
Medical Care Situations.
• Documentation of FDA approval for the Expanded Access Use request
Compassionate use of unapproved drug: this use will require FDA and IRB approvals (or IRB chair
concurrence as explained later) before use. The physician may use
FDA Form 3926 for the FDA
submission. A physician submitting an individual patient expanded access IND using Form FDA 3926
may select the appropriate box on that form to request authorization to obtain concurrence by the IRB
chairperson or by a designated IRB member before the treatment use begins, in lieu of obtaining IRB
review and approval. If the sponsor is submitting an Modification to an existing IND via Form FDA 1571,
and because the form does not have a similar box, the information can be included in a separate request
with the application.
If seeking IRB concurrence, the request should include the following:
• A copy of all information submitted to the FDA in connection with the Expanded Access use request
• Informed consent form to be used
If the FDA does not grant waiver of IRB approval or if the request was not made to the FDA via FDA Form
3926, the study team should submit an IRB application that would be routed to the Full Board per the
regulations.
If the documentation received indicated the FDA has approved a waiver of IRB approval, the Team Q
member will create a folder with the information received from the treatment team. To document the
use in the system, the study team will create an RNI submission and will add all the documents required
for the concurrence. The Team Q member should redact any patient information in the RNI submission
and upload revised redacted copies of documents, if applicable.
The Team Q member will contact the study team in case of any required changes while alerting the IRB
Chair about the Compassionate use. The IRB chair will confirm his/her concurrence with the use of the
drug via e-mail. This concurrence will be sent to the study team. The use can only start after the FDA
approves the Compassionate Use and the IRB concurs. The Q team member acknowledged the RNI.
If the study team is submitting to the IRB instead of asking IRB chair concurrence, or if the IRB chair
decides the use should be reviewed via Full Board, the study will be routed as usual via a new study
submission.
REFERENCES
• FDA webpage: Expanded Access (Compassionate Use) at
https://www.fda.gov/NewsEvents/PublicHealthFocus/ExpandedAccessCompassionateUse/default.h
tm#Investigational_Drugs

Table of Contents
Page 86 of 273
• FDA Guidance: Expanded Access to Investigational Drugs for Treatment Use — Questions and
Answers at https://www.fda.gov/downloads/drugs/guidances/ucm351261.pdf
• FDA Expanded Access for Medical Devices Guidance at:
https://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/Inv
estigationalDeviceExemptionIDE/ucm051345.htm
• FDA Presentation: Emergency Use and Compassionate Use of Unapproved Devices at
https://www.fda.gov/downloads/Training/CDRHLearn/UCM180888.pdf
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
1/31/2018
Revamp of process per FDA changes; adding drugs to SOP
3/4/2020
Change in the process to align with the new system
12/11/2020
Added that PHI should be redacted from documents

Table of Contents
Page 87 of 273
SOP Title: External Webinars presented by Team Q
SOP Category:
QA and Education
Established:
1/23/2018
Last Revision:
8/15/2024
PURPOSE
The purpose of this document is to explain the process Team Q members will follow to schedule an
external webinar for our staff, ORA/OC staff or IRB members
SCOPE
The SOP applies to external webinars presented by the Q team
RESPONSIBILITIES
• QA IRB analyst – if applicable, make sure to obtain a sign in information for attendees and scan it to
the designated folder in the H drive
• Q IRB Team Supervisor-schedules the webinar and emails potential attendees.
• AVP or IRB Director- may schedule a webinar and indicates if a webinar is required for the IRB Staff
PROCEDURE
• The AVP or IRB Director or Q team member (delegated the taskper AVP or Director) will purchase an
external webinar. The IRB Director will let Team Q members know if staff attendance/viewing is
mandatory
• The Q IRB Team member will schedule the webinar, finding a room suitable for the presentation and
or scheduling via Zoom . The information will be emailed to the attendees. This list may include
other IRB staff, IRB members, or staff from other divisions (at the director’s request). The email
should contain:
o Link to the webinar information
o If the webinar offers credits and, if so, how to obtain those
o Copies of the PowerPoint for the presentation, if available
• The registration information will be saved in a folder under the external webinar folder in the Emory
IRB H drive. This can be found at H:\General\Education\External Webinars
• If allowed, any room reserved for the webinar should be reserved from 15 to 30 minutes before the
webinar start time to allow for set up. This reservation will be sent only to Team Q members
• On the day of the presentation, the QA IRB analyst will capture all attendees. If not in person, the
analyst should keep their certificate of attendance when available and forward it to their supervisor
when requested.
• After the webinar is finalized:
o If applicable, the QA IRB analyst will scan the sign-in page and save it under the external webinar
folder that corresponds to the webinar. This can be found at H:\General\Education\External
Webinars.
o The Q team member will send the powerpoint presentation (if not sent earlier), and an
attendance certificate/survey if applicable.

Table of Contents
Page 88 of 273
PROCESS FLOW
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/25/2021
Updates to reflect remote working arrangements
8/15/2024
Further updates for remote considerations
IRB
Director/Team
Q purchase
webinar
Attendees are
notified of
details
Team Q
member
creates sign in,
and after
webinar, scans
completed
form to H
drive folder
Team Q emails
attendees
certificate of
attendance
and slides as
applicable

Table of Contents
Page 89 of 273
SOP Title:
Process of Review of Consent Process Errors after Notification from OCR
SOP
QA and Education
Established:
1/2/2014
Last Revision:
1/2/2014
PURPOSE
The purpose of this document is to explain the process the IRB Team Q designee will follow after
receiving an email from the Office for Clinical Research (OCR) about the use of an expired consent form.
SCOPE
The SOP applies to Emory studies that meet the NIH definition of Clinical Trial with or without EHC
billable items and services, AND clinical research which doesn’t meet the definition of Clinical Trial, but
does have EHC billables items and services. For these studies, a copy of the informed consent form (ICF)
is placed on the electronic medical record by OCR.
DEFINITIONS
• Expired ICF: ICF that has an expiration date in the header of the document that precedes the date
the subject signed the ICF. An expired ICF is not necessarily equivalent to an unapproved ICF
version.
• ICF version: the number or date in the footer of the document. Any changes to the ICF’s content,
approved by the Emory IRB, are reflected in a new ICF version date.
RESPONSIBILITIES
• IRB OCR representative – emails the IRB Team Q designee and the study team about the use of an
expired ICF in a study approved by the Emory IRB. This person also will upload the ICF, and letter
from IRB to the Emory electronic medical record (EeMR) if appropriate.
• IRB Team Q designee- reviews information, and emails OCR and study team as appropriate.
PROCEDURE
• When OCR receives an expired ICF via fax, email, or hardcopy, the OCR representative will email
the study team to let them know that the consent may not be acceptable, copying the team Q
designee and the IRB Director.
• The Team Q designee checks the ICF to see if the version of the ICF is that which is currently
approved by the IRB.
• If the expired ICF is the current version:
o Following the OCR email to the study team, the Team Q designee will reply-all stating this,
and asking the study team to have the subjects sign an un-expired copy of the ICF.
o The Team Q designee will also ask the study team to submit a reportable new information
submission if any procedure took place before the subject reconsented.
o In addition, the Team Q designee will email a letter to OCR stating that the expired ICF is the
version currently approved by the IRB (See attachment 1).
o OCR can then, at their discretion, upload the expired ICF into the medical record and the
letter from the IRB.
• If the expired ICF is not the currently approved version:
o The Team Q designee will reply-all stating the expired ICF is not the currently approved
version, and will ask the study team to reconsent the subject(s) as soon as possible, and to
send the signed correct ICF(s) to OCR to upload to the EeMR.

Table of Contents
Page 90 of 273
o The Team Q designee will also ask the study team to submit a reportable new information
submission if any procedure took place with the affected subject(s) before they were
reconsented. The information will be sent to the CoRe team per the IRB’s current process if
applicable.
o If OCR receives the correct ICF forms, a letter will not be necessary.
PROCESS FLOW
Process flow when the ICF version used is the same one approved by the Emory IRB
Process flow when the ICF version used is not the same one currently approved by the Emory IRB
Attachment 1
* Use IRB letterhead for this letter. Delete instructions in red.
OCR emails
study team and
Q designee
Q designee
emails back
with
reconsenting
intructions, ask
study team to
submit RE if
procedures took
place and sends
letter for OCR
OCR will upload
either expired
ICF in eEMR
with the IRB
letter, or
unexpired
consent if
reconsent was
possible
If procedures
took place
before
reconsent, the
Q designee will
send event to
designated
reviewer
OCR emails study team and Q
designee
Q designee emails back asking
study team to reconsent and to
indicate if procedures took place
prior to reconsent
OCR will not upload ICF in eEMR and
will wait to receive correct ICF
version after reconsent
If OCR receives the correct ICF from
the study team, they will inform the
Q designee
If activities took place, Q designee asks
study team to submit a RE. The RE
case manager sends event to CoRe.
The Q case manager sends letter to OCR
indicating the CoRe (FB) findings, final
determination, and CAPA plan for the
study, if OCR did not receive the subject
reconsent form

Table of Contents
Page 91 of 273
DATE
Office for Clinical Research
RE: IRB#_______; PI: NAME
To whom this may concern,
On DATE we received an email from your office indicating an issue during with a subject’s informed
consent process documentation (list the name of the subject; it should be one subject per letter for
privacy requirements when uploading to the EeMR). Specifically, DETAILS OF EMAIL. During our
review, we found the following:
The informed consent form (ICF) used to consent this subject has the same content as the
currently IRB approved version of the ICF. We have instructed the study team to reconsent the
subject for documentation purposes, and to submit a reportable new information submission if
any research activities took place before the subject was reconsented.
You may use this letter for documentation purposes. Please let us know if you have any
questions.
Sincerely,
NAME
TITLE
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 92 of 273
SOP Title:
Informed Consent Monitoring SOP
SOP Category:
QA and Education
Established:
2/24/2010
Last Revision:
2/24/2010
PURPOSE
Representatives of the IRB (generally, members or staff) may conduct real-time informed consent
observations as part of Emory’s human research protection program. This is a valuable measure for
providing constructive feedback to investigators and ensuring the highest quality experience for
prospective research subjects.
AUTHORITY & SCOPE
Federal regulations authorize an IRB to observe or have a third party observe the consent process and
the research. The IRB staff will conduct informed consent monitoring in studies under the Emory IRB
oversight.
PROCEDURES
• The IRB staff will give study teams advance notice of a proposed informed consent monitoring visit.
• The Emory IRB Informed Consent Monitoring Checklist should be used to capture relevant
information for each session.
• The following procedure should be followed for consent observation:
o In a private room or waiting area, the person obtaining informed consent should tell the
prospective subject that a representative of the IRB is on site to observe the informed consent
discussion. The IRB representative should then introduce him/herself to the prospective subject
and ask for permission to observe the consent session. If the subject declines, the IRB
representative will not observe that consent discussion.
o The person obtaining informed consent will then conduct the informed consent discussion as
usual, while the IRB representative observes silently, without taking notes or interjecting into
the discussion.
o The subject should be free to ask the IRB representative questions. If this happens, the IRB
representative should answer them in a helpful manner.
o At the conclusion of the discussion, the IRB representative will thank the subject and person
obtaining informed consent and will leave the room. At that time, s/he should complete the
checklist.
o If the study team requests it, the IRB representative may may give immediate feedback on the
discussion and mention any deficiencies in the process.
o The IRB representative will send the PI a letter in follow up describing the deficiencies from the
observation, if any, within three business days. Recommendations for improvements should be
included in this letter. Study teams are encouraged to discuss the findings with the IRB
representative or IRB Director.
o If deficiencies in the consent process are noted, the IRB staff member may refer those findings
to the IRB Compliance Review team, require additional education on the consent process, or
provide on-site informed consent training for the study staff.


Table of Contents
Page 94 of 273
SOP Title:
Internal QA/QI review of documents after IRB Review
SOP
QA and Education
Established:
6/1/2012
Last Revision:
8/15/2024
PURPOSE
The purpose of this document is to describe the process of auditing studies reviewed by the Emory IRB
members and staff.
SCOPE
The SOP will apply to human subject research studies reviewed by the Emory IRB.
OBJECTIVES
• To ensure that the review categories are correct
• To verify that the IRB staff is meeting the turnaround time targets
• To make sure the documents issues by the IRB are correct (including dates, information such as
device risk determination, ICF/HIPAA, etc.)
• To verify that the Emory policies and procedures, and federal regulations are followed during study
review, including studies under Department of Education, Department of Justice, Department of
Defense, Department of Energy and the Environmental Protection Agency.
RESPONSIBILITIES
• IRB QA Team member – reviews determinations made by IRB staff and IRB members, and minutes
that applied to the item, if applicable. In addition, the QA team may review items prior to review
assignment. IRB leadership may participate in these activities.
• IRB staff- The analyst will correct the documents, after confirmed findings from an audit which are
corroborated by the IRB Associate or Assistant Director or AVP. Analyst also handles proper
archiving of corrected documents.
• IRB Full-Board- reviews comments and approves corrected documents/minutes, if applicable.
• Designated reviewer- approves corrections of otherwise approved information, if applicable
• Full Board designated reviewer: an experienced person from our staff team who will review studies
that were reviewed at Full board after the minutes are approved.
PROCEDURE
On an ongoing basis, at least monthly, team Q will review a sample of expedited or Full Board new
studies, submitted to the IRB office.. The review will aim to include at least one item per study analyst.
The Team Q member will populate a dedicated form for this purpose. The findings will be conveyed to
the IRB analyst and supervisor. If necessary, the study team will be contacted for other follow up
measures.
During the review, team Q will review the following documents, at a minimum:
• IRB Approval letters, if review complete
• Electronic study history
• Informed consent and HIPAA forms.
• Review of the correct use of review categories, subparts (for vulnerable populations), consent
waivers, and applicable regulations.

Table of Contents
Page 95 of 273
The QA team will verify that submissions contain required information, as applicable (e.g. required
templates, device risk determination, etc), and that the informed consent and HIPAA authorization and
or waiver worksheets have all required elements.
For Full Board studies, a designated person will review a set of full board minutes, and will ensure the
following:
• IRB determinations after study approval, or pending/deferred, are adequately addressed in the
letter to the study PI and resolved, if applicable
• IRB-approved consent/assent forms are stamped correctly
• Other issues as applicable.
PROCESS FLOW
REFERENCES
• IRB policies and procedures
• 45 CFR 46.101
• 45 CFR 46.102
• 10 CFR 745
• 34 CFR 98.3
• 28 CFR part 46
• 28 CFR part 512
• 40 CFR 26 Subparts C and D
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/13/2015
This SOP replaced Audit of Expedited, Exempt and Non-Human Subject Research Determinations
and Internal QA/QI review of documents after IRB Full Board. Major overhauled to document the
current review process
11/1/2017
Changes to the subject matter reviewed, as ADs are reviewing FB studies at the moment of
auditing the FB minutes
6/9/2021
Added additional details for the review of full board studies
8/15/2024
Clarified updated QA procedures
QA Team Member
reviews assigned Full
Board new studies
and a subset of
expedited studies
meeting certain
criteria
FIndings are relayed
to analyst and
supervisor
If necessary, the
study team will be
contacted to correct
further.

Table of Contents
Page 96 of 273
SOP Title: Letters after FB with PIs, OHRP and FDA after SNC, CNC and UP determinations
SOP
Category:
QA and Education
Established:
5/15/2013
Last Revision:
9/6/2013
• Letters to the PI and federal oversight agencies will be drafted by the case manager.
• Any letter to inform PIs about SNC, CNC or UP determinations should be reviewed by another
member of Team Q for accuracy and readability before sending.
• Any letter to notify the FDA or OHRP about a SNC, CNC or UP determinations should be reviewed by
the AVPP of the HRPP or their delegate before sending.
• For notifications to the FDA, the letter should be signed by the IRB chair (who is a MD or clinician).
In the case of an absence of the IRB chair, a MD vice-chair may sign on the IRB chair’s behalf. For
notifications to OHRP, the AVPP of the HRPP or their delegate should review the content of the
online submission. In case the AVPP of the HRPP or their delegate is absent, the a co-chair or the
Institutional Official could review the submission details.
• Once the letter has been signed, the letter should be scanned as a PDF document and saved in
H:\irb_shared\General\QA Working Files\CMTE Q\YEAR\DATEofMTG\Letters to Feds
• All letters should be saved in their respective location for archiving purposes.
• The electronic copy of the letter to the PI should be emailed to the PI and Institutional officials, per
our guidance document entitled: Everything you need to know when writing letters to PIs, FDA and
OHRP after a FB determination of SNC, CNC or UP. A copy of this email should be kept under
H:\irb_shared\General\QA Working Files\CMTE Q\YEAR\DATEofMTG\Letters to PIs.
• For letters to FDA/OHRP, use guidance document entitled: Everything you need to know when
writing letters to PIs, FDA and OHRP after a FB determination of SNC, CNC or UP. See Contact Grid
for IOs who should be copied and Fed (FDA, OHRP) contacts. When sending the email to the federal
agencies:
o Email the FDA by themselves
o Forward that sent email to the PI, copying the IOs
o Save a copy of the email to the PI (that also contained the information of the email to the
federal agencies) under H:\irb_shared\General\QA Working Files\CMTE
Q\YEAR\DATEofMTG\Letters to Feds
• If you receive an acknowledgment of receipt from OHRP or FDA, file it in the appropriate folder.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
10/14/2022
Updated some of the process steps

Table of Contents
Page 97 of 273
SOP Title:
Reportable new information submission Review Process
SOP
QA and Education
Established:
7/29/2013
Last
02/24/2023
PURPOSE
The purpose of this document is to describe the review process of reportable new information (RNIs)
submitted to the Emory IRB office.
SCOPE OF SOP
This SOP scope is all RNIs submitted to the Emory IRB.
SOP SECTIONS
Screening of RNIs
Events sent to the CoRe team
Events sent to Committee Q
Cases involving a UP decision that affects multiple studies
DEFINITIONS
• IRB Team Q: Specialized IRB staff who reviews reportable events, work on fact-finding with study
team, and submit events to a designated reviewer, Compliance Review (CoRe) team and/or
Committee Q as applicable
• Designated Reviewer: A member who has been designated by the Chair to perform expedited
reviews on a term basis, or as needed case by case, preferably in writing.
• CoRe Team: A designated group of the IRB Chair, Director, and qualified IRB staff that reviews
reported events (including alleged non-compliance, potential UPs, potentially serious or continuing
non-compliance), suspensions, and terminations. The CoRe team triages cases to determine
whether they need a review at a convened meeting of the Emory IRB. The CoRe may engage the
assistance of ad hoc consultants.
• Committee Q: A Full board meeting that primarily reviews potential serious and/or continuing
noncompliance and unanticipated problem cases.
• Non-reportable event: An event that is not reportable to the IRB per the IRB Policies and
Procedures.
REPORTS TO AAHRPP
Within 48 hours but as soon as possible after the organization (or individual researcher) becomes aware,
the IRB must report to AAHRPP:
o Any negative actions by a government oversight office, including, but not limited to, OHRP
Determination Letters, FDA Warning Letters, FDA 483 Inspection Reports with official action
indicated, FDA Restrictions placed on IRBs or Investigators, and corresponding compliance actions
taken under non-US authorities related to human research protections.
o Any litigation, arbitration, or settlements initiated related to human research protections.

Table of Contents
Page 98 of 273
o Any press coverage (including but not limited to radio, TV, newspaper, online publications) of a
negative nature regarding the Organization’s HRPP.
Work with the Team Q Lead or the IRB Director to assist with reporting.
PROCEDURE
Screening of RNIs
The Senior Q team member will screen the submission.
• If the case reported is potentially noncompliance, a reportable protocol deviation, or an
unanticipated problem, it will be assigned to a Team Q member (see next section on CoRe Review).
• If the case is considered a minor protocol deviation, the Senior Q team member may send the RNI to
a designated reviewer. In some cases, the RNI may be acknowledged by a designated member
per
SOP.
• Other cases sent for Acknowledgment: If there is precedent for a case, via CoRe and/or FB, that
indicates that the RNI is potentially noncompliance, but neither serious nor continuing, the Senior Q
member may send the RNI for expedited review. The following specific precedents are already in
place for sending to expedited review:
o Noncompliance involving the data collection from more charts than what was approved by the
Emory IRB.
o Over-enrollment of subjects, in cases where the study is expected to enroll a large number of
subjects as, for example, in a multi-site trial. If the study is considered of higher risk (for
example a Phase I study) or the study has over-enroll with more than 25% of the approved
number, the RNI should be sent instead to the CoRe Team.
• To send to a designated reviewer
o Complete the pre-review and select “Additional review required” along with other applicable
information.
o The review can then be assigned to the designated reviewer for processing
o If you are not sure if the case needs to go to CoRe, ask the designated reviewer to weigh in and
determine if it should be acknowledged versus sent to CoRe. If they want to route the RNI to
CoRe, ask them to email you instead without completing the review.
o Once the review is submitted, this completes the acknowledgment process. A letter is not
required in these cases; however, it is a best practice to log a comment to the team informing
them of our process for managing acknowledgments.
Events sent to the CoRe team – see contact grid
for who should be included in CoRe email.
• Create a folder on the H drive in the QA Working Files\Non-Compliance or UP folder with study title
and PI name, using the following format (RNI IRB/STUDY##PILASTNAME plus initials of case
manager).
o When the case is under review, it should be located under the Pending folder.
o If closed, the case should be moved to the closed folder. It should be located at H:\General\QA

Table of Contents
Page 99 of 273
Working Files\NC UP Complaints\NonCompliance\Closed\ RNI IRB/STUDY##PILASTNAME
removing the case manager initials.
• The Team Q member should contact the PI and study team for the input and clarification of data,
corrective and preventive action plan, or any other information if needed. Use current SOP Ed & QA
Team Mission and Process.
• The information obtained is added to the worksheet. The form can be located at: H:\General\QA
Working Files\Forms, templates and Guidance\ Forms for RNI review
• The team Q member emails the worksheet information to the CoRe team. The information from the
form should contain information from the title of the study to the CAPA plan. Other appropriate
documents may be attached as well.
• The CoRe team as of June 14, 2021, , is composed of Carlton Dampier, Cliff Gunthel, Shara
Karlebach, Julie Martin, Rebecca Rousselle, Aryeh Stein, and Larry Tune.
• The CoRe team should “Reply All” via email with the recommendations. If anyone on the CoRe team
thinks the case could represent serious or continuing noncompliance (SNC/CNC) or an unanticipated
problem (UP), refer the case to the next Com. Q meeting. If three CoRe members think the case is
not SNC, CNC, or UP, including at least one clinician, or at least one Vice-chair or Co-Chair, the case
can be closed and you can prepare the letter. Also, if any member of the CoRe has a conflict, they
should recuse him/herself from the review of the RNI.
• The cases will be divided into Clinical CoRe cases and Procedural CoRe cases.
o Clinical CoRe cases may be those involving clinical care or participant safety such as a missing
clinical test or dosing issues..
o The Procedural CoRe cases may be those that are nonclinical in nature such as HIPAA issues,
protocol deviations, noncompliance and unanticipated problems not involving a clinical matter.
o Team Q may decide at their discretion to whom to send a case, but members will be selected
according to their area of expertise. In cases that involve both clinical and nonclinical issues,
route it to clinical CoRe. Only 5 CoRe members will be selected for each case, always including a
Co-Chair and the three staff members.
• If the CoRe team determines that the RNI represents no noncompliance, not serious or continuing
noncompliance or not an unanticipated problem, the RNI will be closed. The RNI worksheet should
be completed and non- applicable sections should be deleted.
o The RNI should be closed in eIRB by submitting the RNI pre-review with the appropriate
determination. Once the pre-review is submitted, the case manager should still send a letter to
the study team in the system.
o Some pre-review determinations will automatically move the RNI to “acknowledged” status, but
a letter should still be written for cases closed by CoRe. You may have to navigate to find the RNI
if it moves from your inbox as a result of the pre-review determination.
• If CoRe has determined that a safety report submitted in an RNI is not a UP, additional studies
reporting the same safety event can be sent in an abbreviated form to the CoRe, with information
about the study, to confirm that applies to this new study. The email should be saved in the folder
on the shared drive in lieu of a worksheet. The RNI letter should also be saved in the same folder.
• If the case is closed after a CoRe determination, the folder should contain the following:

Table of Contents
Page 100 of 273
o Letter to the PI: study number, PI last name, Letter to PI, date of issue (example 123 Smith Ltr to
PI 11-5-12
o CoRe correspondence
o Completed RNI worksheet
Events sent to Committee Q
• If the CoRe assesses the RNI as potentially SNC or CNC, the RNI should be forwarded to committee Q
for review.
• If the CoRe assessed the information as a potential UP, the RNI should be sent to the next available
full board meeting or CMTE Q if the RNI does not represent an immediate safety issue for subjects.
• The following actions should be completed:
o Submit RNI Pre-Review
Select additional review required to assign to the Q meeting
o The case manager is also required to inform and forward the invite CHOA or VA colleagues as
per the contact grid of any cases going to CMTE Q involving CHOA or VA facilities via email. VA
cases are required to be reviewed within 30 days.
o Save the materials in the appropriate CMTE Q folder (do not include CoRe emails).
o Contact the PI to schedule the SNC/CNC/UP review for a Committee Q meeting. The PI must be
invited to attend by zoom, in person, or phone. Attendance is only required for compliance
reviews. If the PI or a PI representative cannot attend, the study team may submit a response in
writing. Schedule the case for an agenda based on the PI’s availability. If the PI cannot come to
the meeting, consult with CoRe about moving the case to the next available meeting. In the case
of UPs, the case should be reviewed at the next available meeting if it represents imminent
harm to subjects, despite PI availability.
• If the board determines that an RNI constitutes an unanticipated problem or serious or continuing
noncompliance, some or all of the following actions could be required:
• Suspension of the research
• Termination of the research.
• Notification of current participants when such information might relate to participants’
willingness to continue to take part in the research.
o Modification of the protocol.
o Modification of the information disclosed during the consent process.
o Providing additional information to past participants.
o Requiring current participants to re-consent to participation.
o Modification of the continuing review schedule.
o Monitoring of the research.
o Monitoring of the consent process.
o Referral to other organizational entities.
After the meeting

Table of Contents
Page 101 of 273
• Team Q will meet to review findings from the meeting and submit committee reviews by adding the
worksheet to the RNI Committee Review information under question 5. During this time, the CAPA
plan will be added as well per the IRB Team Q CAPA Follow Up SOP, if applicable
. Question 6 should
be marked as yes if you are ready to finalize.
• The PI should receive a determination letter within 2 business days. Follow available templates and
make sure institutional officials are copied per the contact grid if the issue was determined to
represent an UP, SNC or CNC by the convened IRB. For external UPs, follow the contact grid for who
should be copied.
• If applicable (after a UP, SNC or CNC determination), a letter to OHRP and FDA should be drafted
and sent to the AVP for the HRPP for review (refer to SOP). Specifically:
o For studies funded with federal funds: report to OHRP
o For studies using an FDA regulated product: report to FDA
o If both of the above apply, report to both. You may be able use a PDF of the OHRP online
submission to the FDA.
• Once the FDA/OHRP letters/online submission have been reviewed by the AVP for the HRPP, they
can be reported to the applicable Fed agencies. Once that reporting is complete, email the PDF of
the online submission or forward the FDA email to the study PI and university officials as per the
contact grid.
• The letters to OHRP and FDA should be logged in a comment under the RNI history and saved in the
RNI folder. These letters should also be saved under the CMTE meeting folder entitled “Letter to
Feds.”
• The RNI is closed in eIRB by sending the letter to the study PI. Once the case is considered closed,
the RNI folder should be archived in the closed folder.
• Save a copy of the sent email correspondence under “Letter to Feds” folder
• If the case is closed after a FB determination, the folder should contain the following:
o Letter to the PI: study number, PI last name, Letter to PI, date of issue (example 123 Smith Ltr to
PI 11-5-12
o CoRe correspondence: CoRe determination on DATE
o Copy of worksheet
o Copy of Letter to Feds: study number, PI last name, Letter to FDA or OHRP, date of issue
(example 123 Smith Ltr to FDA 11-5-12)
• If the case has a CAPA pending item (RNI Action), the case should be moved from the “pending”
folder to the “working on CAPA” folder. When the CAPA issue (RNI Action plan) is completed, the
case can be moved to the “closed” folder.
For AVAMC Research:
• If the convened IRB or the IRB reviewer/CoRe determines that the problem or event was serious,
unanticipated, and related to the research, a simultaneous determination is required regarding the
need for any action (e.g., suspension of activities; notification of participants) necessary to prevent
an immediate hazard to participants in accordance with VA regulations.
o All determinations of the IRB reviewer (regardless of outcome) must be reported to the IRB at
its next convened meeting.

Table of Contents
Page 102 of 273
• The VA medical center director must report the problem or event to the appropriate VA Office of
Research Oversight research officer within five business days after receiving such notification.
• If it was determined that the problem or event is serious, unanticipated, and related to the research,
the convened IRB must determine and document whether a protocol or consent document
modification is warranted.
• If the convened IRB determines that a protocol or consent document modification is warranted, the
IRB must also determine and document:
o Whether previously enrolled participants must be notified of the modification.
o When such notification must take place and how such notification must be documented.
Cases involving an UP decision that affects multiple studies
• During the meeting, the board may make a UP determination. If the determination affects studies
using the same drug or device, the board should make a note on the meeting minutes stating one or
more of the following options:
o IRB case manager informs other study teams that are using that drug or device. Study teams
should review the information for the UP, and assess if an RNI and a MOD if applicable, is
needed to add the new risk to their study documents.
The study team may disagree, and if so, they should document why this UP does not affect
their study.
The RNI and MOD submission can be reviewed via the expedited process as a risk/benefit
ratio analysis was done during the convened IRB meeting.
• After the meeting, the case manager will send an email to the IRB staff, letting them know that this
UP may affect several studies, and that the RNI and MOD can be reviewed expedited, if applies. The
email should contain the list of the studies.
• In addition, the case manager will log comments to each study, asking the study team to submit an
RNI and a MOD as applicable. If the original RNI had letters from the sponsor explaining the issue,
they should be added to the comment. Here is an example for this comment:
Dear study team,
The IRB received the attached letter in a reportable new information submission involving a
different IRB study using the same investigational agent than your study. The reportable new
information was reviewed at an IRB convened meeting on DATE, and determined that the event
of NAME OF EVENT, represents an unanticipated problem.
The Board determined that this UP determination may affect your study. As such, you are may
be required to submit a reportable new information. Please review the attached document, and
submit a reportable new information with your assessment of this risk. If you do not agree with
the submission of the reportable new information please let us know why this event does not
apply to your study population. If you agree with the UP determination, you may also be
required to submit an amendment to add the risk to the informed consent form. If this risk is
already in your consent form, you may disregard.
Your reportable new information and amendment will be reviewed via expedited process, as
approved by the full board meeting on DATE. You have 10 business days to submit the RNI and

Table of Contents
Page 103 of 273
AM or provide your assessment of why this event does not apply to your study. If your study is
reviewed by an external IRB, please report to them directly.
Let us know if you have any questions,
• The case manager is responsible to check that the RNIs and MODs have been submitted if required.
The MODs will be processed by the study owner. If the study team does not take action in the
allowed time, the case manager needs to escalate emailing the study team and the Senior Q
member with the following information:
Dear Dr. X,
On DATE, you were notified of an unanticipated problem determination that may affect your study.
You were required to get back to us and indicate if this determination negatively impacts your study.
Please, respond to this email with your assessment by close of business on DATE (Friday of this
week). Failure to respond by this dateline represents noncompliance.
If you have any questions, please let us know,
NAME
• When the RNI is submitted, the information can be sent to a designated reviewer (DR) for expedited
process. The DR should be a member of CMTE Q members who is a medical doctor. The message to
the DR should include this information:
o Determination was made during CMTE X on DATE
o Assess if the UP determination also affects this study
o Attach copy of the meeting materials and determination letter
o Here is an example of the information for DRs:
Dear Dr. X,
This same event was reviewed by CMTE X on DATE for another study (IRB 123456). The event,
NAME OF EVENT, was considered an UP. This same event is being reported for several studies.
We need your help assessing if this event also applies for this study. If so, we will acknowledge
this event as an UP for this study as well. Please submit an RNI to document the event. If you feel
this event may affect this study differently and needs to be assessed again, let me know and I
will send to the CoRe team.
Looking forward to hearing from you,
NAME of TEAM MEMBER
PS: See attached documents from the Q meeting for your reference
• After the DR reviews the RNI, the RNI may be acknowledged or sent to CoRe for new review (if the
DR does not think the UP determination affects this study). If the latter, the RNI will follow the CoRe
review process as outlined before.

Table of Contents
Page 104 of 273
• If the RNI was confirmed as an Unanticipated Problem by the DR, the case manager will close the
RNI with a letter to the study team.
REFERENCES
• IRB policies and procedures
• Emory SOP: Acknowledgments & Noncompliance Determinations Made by Senior Team Q Staff
• Emory SOP: Letters after FB with PIs, OHRP and FDA after SNC, CNC and UP determinations
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/25/2021
Adding required reporting to AAHRPP. Clarified reporting lines.
5/24/2021
Adding a step for cases routed to a DR, changing CoRe composition starting on June 14, 2021.
7/2/2021
Clarified a component of CORE email voting
10/14/21
Revised CHOA contacts and archived updates over 1 year old
1/21/2022
Revised CHOA contacts and revised SaaS steps to move something from CoRe to Committee Q review
4/28/2022
Replaced Meredith Capasse with Anna Lum
02/24/2023
Revised for clarity throughout

Table of Contents
Page 105 of 273
SOP Title: Education and Quality Assurance Team Mission and Process
SOP Category: QA and Education
Established:
9/30/2011
Last Revision:
9/30/2011
MISSION
To support the Emory Research Community via Education/Outreach and effective quality assurance
practice on behalf of the Emory IRB.
PROCESS
When a case is assigned to a member of the Education and QA team, the process will continue as
follows:
If all attempts to obtain information for a case are unsuccessful, the E/QA team member will consult
with IRB Director for next steps.
a) Communication strategies when obtaining information via email.
(a) Always remember to remind the study team that you are a liaison between the
IRB and the study staff and that you are gathering information needed by the IRB
(or IRB members) so they can make a determination.
(b) Have a friendly tone in your email, including words such as “collaborative” or
“working together”, to reflect the cooperative mission of the E/QA team.
(c) Help the study team with their corrective action plan, suggesting plans that
worked for other teams.

Table of Contents
Page 106 of 273
(d) Do not jump to conclusions when asking questions. Remember to obtain
complete information before sending the case to the CoRe team (if it is not
something that could increase the risk for participants or others).
(e) If the information is not effectively obtained via email, call the PI/Study team and
offer guidance and the opportunity for an in-person meeting.
b) Communication strategies when obtaining information via phone conversations.
(a) Remember the tone of your voice and inflection when discussing problems with
the study team. Remember to smile when talking.
(b) Remember to include words that reflect the cooperative mission of the E/QA
team as explained above.
(c) Reassure the study team regarding their case. Explain the steps and, if needed,
give them the definitions for Non-Compliance, Protocol Deviations and
Unanticipated Problems.
(d) If the conversation turns confrontational, avoid making any remarks or use of
words that could sound challenging. Always remember to keep a friendly tone
and explain why the questions are asked (you are a liaison for the IRB).
(e) After the phone conversation has ended, promptly email the PI/Study team with a
summary of the conversation, always offering additional help.
(f) If the information is not effectively obtained, offer to have an in-person meeting.
c) Communication strategies when obtaining information in person
(a) In meetings with PI/Study staff, remember that your body language is very
important. Always shake the hand of your interviewee and look him/her in the
eyes.
(b) Do not cross your arms and always have a neutral expression on your face when
asking questions.
(c) If the answer is surprising, avoid making gestures that could make your
interviewee feel uneasy or uncomfortable.
(d) Make notes without being exclusively focused on note-taking. It is important to
look at the interviewee eyes.
(e) Finalize the meeting by thanking the interviewee for his/her time and giving
him/her information to contact you if additional questions arise.
(f) After the meeting, promptly email the study team with a summary of your
conversations and next steps.
Remember that when dealing with difficult PIs or staff teams, you can always direct the call/email to
the IRB Director for assistance.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 107 of 273
SOP Title:
IRB Record Review of Studies approved by the Emory IRB
SOP
QA and Education
Established:
8/30/2011
Last Revision:
8/15/2024
PURPOSE
The purpose of this document is to describe how the IRB Team Q conducts record reviews of studies
approved at the Emory University IRB. The Emory IRB is committed to helping the research community
maintain a high standard of compliance through education and guidance. Record Reviews will play an
integral role in achieving this goal as it will allow the identification of problems offering opportunities for
education.
SCOPE OF SOP
Record Reviews conducted in human subjects’ research overseen by the Emory University IRB and
studies conducted at Emory under external IRBs.
DEFINITIONS
• Not-For-Cause Compliance reviews – Periodic compliance reviews are conducted using a
systematic method to review IRB-approved research or IRB records and activities on a regular
basis. The IRB will perform 10 routine record reviews per year.
• Directed Reviews – Assigned to Team Q from the IRB, ORA offices, or Emory affiliates to address a
concern or previously identified potential for noncompliance.
• For-Cause Compliance Reviews: Designed to assess the Investigator’s compliance with federal
regulations, state laws, and Emory IRB P&Ps. These reviews of IRB-approved research studies are
in response to credible evidence of identified concerns or alleged noncompliance.
RESPONSIBILITIES
• Principal Investigator – After the IRB has notified the PI of the record review, the PI and study staff
are responsible for providing all the required documentation as described in the notice. The PI is
also responsible for responding to the record review findings within 5 calendar days of receiving
the record review summary.
• IRB reviewers – The reviewers are responsible for giving a report of findings within 10 calendar
days of the completion of the record review. The written report to the PI will include the IRB
reviewers’ findings and a suggested corrective and preventive action (CAPA) plan, along with a
reasonable deadline for completion (e.g. a month)The PI may adopt the CAPA as provided, or
modify it to match his or her needs. The IRB reviewers will schedule reminders for follow up to
ensure compliance
• Reporting: After the report has been reviewed by the PI and the CAPA plan has been accepted, the
IRB reviewer will send the report to the compliance review (CoRe) team. If the CoRe team makes
a determination of possible serious or continuing non-compliance or an unanticipated problem
increasing the risk for participants or others, the report will be reviewed at the full board meeting.
PROCEDURE
The process will start by communicating with the research staff and coordinator about the record review
visit. The notice for the record review may vary according to review type. For Not-For-Cause
compliance reviews, the study team will have two-week (10 business days) notice from the date of the

Table of Contents
Page 108 of 273
initial communication to the date of the record review. For-cause compliance reviews will be conducted
within 2 business days of the IRB’s initial contact with the study team. The study team should respond
within 24 hours to schedule the record review. Directed record reviews will be conducted according to
study team needs or as mandated by the IRB.
1. The IRB will contact the study team to schedule the meeting. The study team has from 1 to 5
business days to respond to this request, depending on the record review type.
2. The IRB will conduct the record review on the scheduled date. The minimum documents needed
for review are the study regulatory binder (for clinical research), protocol (all versions), informed
consent and HIPAA consent forms (all versions), correspondence with the IRB, IRB submission
records and individual study subject information. The IRB reviewers may give real-time feedback
and answer questions about the information that is being reviewed. The IRB reviewer may also
ask questions to the PI and study staff in case of doubts when performing the record review. The
IRB reviewer may not communicate expected actions of the IRB.
3. The IRB reviewer will send the record review report within 10 business days of the record review’s
completion. The PI will review the findings and CAPA, and determine if the findings are accurate
and if he or she accepts CAPA as is, or if he or she would like to modify it.
4. The PI has 5 business days to respond to the reviewer findings.
5. The IRB and PI, and the study team if desired, will meet the following week to discuss the findings
and next course of action including CAPA implementation and education opportunities.
6. The IRB reviewers will send the report and accepted CAPA plan to CoRe team for review, and later
to the full board meeting if needed.
7. If findings required CoRe review, the study team should submit a RNI in 10 business days. If the
study team do not comply with the request, the Team Q member will create the RNI for them and
ask a super user for submission, documenting the communication with the study team about this
requirement.

Table of Contents
Page 109 of 273
PROCESS FLOW
APPLICABLE REFERENCES
• IRB QA Plan 12.15. 08
• 45CFR 46.
• IRB policies and procedures
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/5/2016
Adding RNI requirements if findings need CoRe review.
8/15/2024
Minor clarification around timing
IRB contacts study group to
schedule the record review
Depending on the record
review type, the study team
has between 1 and 5 days to
respond
Record review is performed on
the scheduled date
IRB record reviewer
prepares the report and
sends it to the PI
PI responds in the next 5
business days.
The PI and staff meet with IRB
reviewers to discuss findings
After report review and CAPA
plan acceptance, the report will
be reviewed by CoRe team. Ask
study team to submit a RE
If CoRe team considered the
findings to constitute possible
SNC, CNC or UP, it will be
reviewed by FB

Table of Contents
Page 110 of 273
SOP Title:
Review of Safety Reports submitted by sponsors holding an IDE
SOP Category:
QA and Education
Established:
6/8/2012
Last Revision:
6/8/2012
PURPOSE
The purpose of this document is to describe the process of reviewing progress reports received under 21
CFR 812.150 (b) (5).
SCOPE OF SOP
The SOP will apply to Emory human subject research working under an IDE.
DEFINITIONS
• Investigational Device: means a device, including a transitional device that is the object of an
investigation. An investigational device is permitted by the FDA to be tested in humans but not
yet determined to be safe and effective for a particular use in the general population and not
yet licensed for marketing.
• Investigational Device Exemption (IDE): An IDE allows an Investigational Device to be used in a
clinical study in order to collect the safety and effectiveness data required to support a
Premarket Approval (PMA) application or a Premarket Notification submission to the Food and
Drug Administration.
• Sponsor: A person who takes responsibility for and initiates a clinical investigation. The sponsor
may be an individual or pharmaceutical company, governmental agency, academic institution,
private organization, or other organization. The sponsor does not actually conduct the
investigation unless the sponsor is a sponsor-investigator
RESPONSIBILITIES
• IRB QA Associate or Assistant Director – reviews the progress report submitted by the sponsor
or sponsor-investigator.
• Sponsor o Sponsor-Investigator: at regular intervals, and at least yearly, a sponsor shall submit
progress reports to all reviewing IRB's per 21 CFR 812.150 (b) (5).
• IRB administrative assistant: scans the progress reports, so they can be added to the study
electronic record by the QA Associate or Assistant Director.
PROCEDURE
Once the IRB receives a progress report (different from the annual report that should be submitted at
continuing review), the Team Q Lead will review the report to verify that it does not contain any new
information that should be reported promptly to the board. If the progress report does not contain new
information, the QA Associate or Assistant Director will forward it to the IRB administrative assistant
who will scan it. The scanned progress reports will be saved under H:\General\Scanned Documents\IDE
progress reports from Sponsors. Once scanned, the IRB administrative assistant will return the hard
copies to the Team Q lead. The Team Q Lead will attach the scanned records to the study electronic
history with a comment indicating that the report did not include any new information and requesting
comments from the study staff about the report review. If the report includes new information that
should be reviewed by the full board, the Team Q Lead will send the report to the CoRe per our SOP
entitled Team Q NC-UP process SOP.

Table of Contents
Page 111 of 273
The report may be destroyed after scanning and archiving under the study history.
PROCESS FLOW
REFERENCES
• 21 CFR § 312
• 21 CFR § 812
• IRB policies and procedures
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
IRB receives progress
report from sponsor
holding an IDE
If progress report does
not contain new
information, the Team
Q Lead will be forward
it to Admin staff to be
scanned. If the report
contains new
information, it will be
routed to the CoRe
team
Once scanned, the
Team Q lead will
attach ther report to
the study electronic
record with a
comment indicating
that no new
information was
identified

Table of Contents
Page 112 of 273
SOP Title:
Routing External UPs (FDA Regulated)
SOP
QA and Education
Established:
7/17/2012
Last Revision:
7/17/2012
PURPOSE
The purpose of this document is to describe how the IRB Team Q will route external unanticipated
problems from studies considered FDA regulated.
SCOPE OF SOP
FDA regulated trials conducted in human subjects’ research overseen by the Emory University IRB.
DEFINITIONS
• Investigational Device: means a device, including a transitional device that is the object of an
investigation. An investigational device is permitted by the FDA to be tested in humans but not yet
determined to be safe and effective for a particular use in the general population and not yet
licensed for marketing.
• Investigational Drug or Investigational New Drug: An Investigational Drug or Investigational New
Drug means a new drug or biological drug that is used in a clinical investigations or a biological
product that is used in vitro for diagnostic purposes.
• Investigational Device Exemption (IDE): An IDE allows an Investigational Device to be used in a
clinical study in order to collect the safety and effectiveness data required to support a Premarket
Approval (PMA) application or a Premarket Notification submission to the Food and Drug
Administration.
• Investigational New Drug (IND) Application: An application that must be submitted to the FDA
before a drug can be studied in humans. This application includes results of previous experiments;
how, where, and by whom the new studies will be conducted; the chemical structure of the
compound; how it is thought to work in the body; any toxic effects found in animal studies; and how
the compound is manufactured.
• Sponsor: A person who takes responsibility for and initiates a clinical investigation. The sponsor may
be an individual or pharmaceutical company, governmental agency, academic institution, private
organization, or other organization. The sponsor does not actually conduct the investigation unless
the sponsor is a sponsor-investigator
• Sponsor-Investigator (S-I): means an individual who both initiates and conducts an investigation, and
under whose immediate direction the investigational drug is administered or dispensed. The term
does not include any person other than an individual. The requirements applicable to a sponsor-
investigator under this part include both those applicable to an investigator and a sponsor
• Unanticipated Problem: any unexpected problem related to the Research, including any unexpected
adverse experience, whether serious or not, that affects the rights, safety or welfare of subject or
others or that significantly impacts the integrity of the research data. OHRP considers unanticipated
problems, in general, to include any incident, experience, or outcome that meets all of the following
criteria:
o Unexpected (in terms of nature, severity, or frequency) given (a) the research procedures that
are described in the protocol-related documents, such as the IRB-approved research protocol
and informed consent document; and (b) the characteristics of the subject population being
studied;

Table of Contents
Page 113 of 273
o Related or possibly related to participation in the research (in this guidance document, possibly
related means there is a reasonable possibility that the incident, experience, or outcome may
have been caused by the procedures involved in the research); and
o Suggests that the research places subjects or others at a greater risk of harm (including physical,
psychological, economic, or social harm) than was previously known or recognized.
• Unanticipated Adverse Device Effect (UADE): any serious adverse effect on health or safety or any
life-threatening problem or death caused by, or associated with, a device, if that effect, problem, or
death was not previously identified in nature, severity, or degree of incidence in the investigational
plan or application (including a supplementary plan or application), or any other unanticipated
serious problem associated with a device that relates to the rights, safety, or welfare of subjects
RESPONSIBILITIES
• IRB Team Q member: reviews initial reported events and sends them to CoRe or FB if appropriate.
• IRB analyst: Makes sure study team reports events through a reportable new information
submission (RE) form.
PROCEDURE
• If an external event is called an unanticipated problem by the sponsor (or S-I), then the event will be
reported via RNI (and Mod if necessary).
o If event does not merit changes via Mod (e.g. study is closed for enrollment without active
subjects), Associate or Assistant Director will send event to DR.
o If the sponsor has not requested changes in the study but the event may merit changes, (e.g.
study is still enrolling or have active subjects) the event will be reviewed by CoRe. CoRe may
acknowledge the event/no changes in study or may recommend FB to review the CoRe
suggested changes to study.
o If sponsor or S-I reports events as unexpected and related but does not call it a UP, the event
will go through CoRe/FB as normal.
• If the event is reported only as an Modification, the IRB analyst will request the submission of a RNI
as well.
• The IRB analyst will contact Team Q lead to notify about the Mod. Team Q only needs to know
about Mod involving increased risk (e.g. changes to the ICF involving new adverse event).
• Team Q lead will assign the RNI to a Team Q member who will be in close communication with the
IRB analyst.
• When the RE/Am goes to FB, the FB should make one of these two determinations:
o The IRB acknowledges the UP determination, and determines that changes as submitted are
appropriate.
o The IRB acknowledges the S or S-I determination, but thinks additional changes are necessary.
• After the FB meeting, the IRB analyst will notify Team Q of the UP determination.
• The Team Q member will send FB determination letter to the PI, copying IOs per contact grid.
PROCESS FLOW

Table of Contents
Page 114 of 273
REFERENCES
• IRB QA Plan 12.15. 08
• 45 CFR 46.103
• IRB policies and procedures
• 21 CFR 812.3
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
RE/Am is received. If only
reported in Mod and risk
may be increased, IRB
analyst request RE
If S or S-I calls event a UP but
does not ask for study changes
•No changes needed-- send to DR
•Changes may be needed-send to
CoRe/FB
If event goes to FB, FB may:
•Acknowledge UP determination and
approve changes
•Acknowledges UP but require
additional changes
IRB analyst (AM owner)
will inform Team Q of FB
action
UP letter done by Q, cc IOs
per contact grid
AM letter done by IRB analyst

Table of Contents
Page 115 of 273
S-I SUBMISSION MANAGEMENT
Definition for the SOPs on this Section
• Investigational New Drug (IND) Application:
a request for authorization from the Food and Drug
Administration (FDA) to administer an investigational drug or biological product to humans which
must be secured prior to administration of any new drug, biological product that is not the subject
of an approved New Drug Application, or Biologics/Product License Application. An IND may be
required for a clinical investigation using a marketed drug for a use other than the indications in the
approved labeling. [21 CFR § 312.3].
•
Investigational Device Exemption (IDE): An IDE allows an Investigational Device to be used in a
clinical study in order to collect the safety and effectiveness data required to support a Premarket
Approval (PMA) application or a Premarket Notification submission to the FDA.
Investigational use
also includes clinical evaluation of certain modifications or new intended uses of legally marketed
devices. All clinical evaluations of investigational devices, unless exempt, must have an approved IDE
(either abbreviated or issued by the FDA) before the study is initiated. [21 CFR § 812.3].
• Sponsor: A person who takes responsibility for and initiates a clinical investigation. The sponsor may
be an individual or pharmaceutical company, governmental agency, academic institution, private
organization, or other organization. The sponsor does not actually conduct the investigation unless
the sponsor is a sponsor-investigator. [21 CFR § 312.3, 812.3].
• Sponsor-Investigator: an individual who both initiates and conducts an investigation, and under
whose immediate direction the investigational drug is administered or dispensed. The term does not
include any person other than an individual. The requirements applicable to a sponsor-investigator
under this part include both those applicable to an investigator and a sponsor. [21 CFR § 312.3,
812.3].
• Annual or Progress Report Submission: A report submitted to the FDA by the IND or significant risk
IDE holder within 60 days of the anniversary date that the IND went into effect or at regular
intervals and at least yearly for an IDE for a significant risk device. The report contains the study
progress information, general investigational plan for the following year, and other applicable
information.
[21 CFR § 312.33, 812.150].
Scope for SOPs in this Section
FDA regulated studies, involving an Emory faculty member holding an IDE for a significant risk device or
an IND.
References for SOPs in this Section
• 21 CFR § 312
• 21 CFR § 812
• IRB policies and procedures
Emory Sponsor Responsibilities Checklist Continuing Review Update=
SOP Title:
New study screen process for S-I studies
SOP
S-I Submission Management
Established:
7/26/2012

Table of Contents
Page 116 of 273
Last Revision:
8/15/2024
PURPOSE
The purpose of this document is to describe the review process of new study submissions by the IRB
analyst and when the study involves an Emory Sponsor or an Emory Sponsor-Investigator (S-I) holding an
IDE for a significant risk device or IND.
RESPONSIBILITIES
• IRB analyst – verifies that the information in eIRB, including the protocol and DSMP, is complete,
requests the S-I or Sponsor complete required S-I training.
• Emory investigator holding IND or significant risk IDE: makes sure that the information submitted in
eIRB is complete and accurate, according to the study protocol submitted to the FDA and complete
required S-I training.
PROCEDURE
• The IRB analyst will direct the study team to add “S-I” to the beginning of the short title (if not
already added).
• The IRB analyst will notify the Sponsor/S-I to complete the required S-I training and ask Sponsor/S-I
to provide additional information about the study IND/IDE (e.g. IND/IDE submission date, FDA
correspondence, Study May Proceed letter etc.)
o If the investigator is applying for a new IND/IDE, a study cannot be placed on a Full Board
agenda until we receive either FDA “study may proceed” correspondence or more than 30 days
have passed since the date of IND/IDE submission. If the team pushes back, the analyst will
contact the Associate Director for additional steps.
• If the investigator is submitting an IND Amendment to add a study under an existing IND, a study
cannot be placed on a FB agenda until documentation that the IND amendment has been submitted
is received. If available, the team should provide FDA correspondence related to the review of the
IND amendment; however, the submission package sent to FDA should be provided. There may be
situations where the IRB requires feedback from the FDA before moving forward with review.
• If the study team received an IND or IDE letter stating that it is pending some contingencies before
proceeding, the IRB analyst will ask the team for the information received from the FDA (via email,
for example) and the changes to the protocol and consent forms sent to the FDA to address said
contingencies. The FDA correspondence should be uploaded under the drug section, question 4
(protocol/ICF changes should be uploaded in their respective section in eIRB). When the FDA has
issued non-clinical hold comments, the IRB analyst will request a response from the team on how
they will address the comments (a copy of correspondence with the FDA is not required).
• When the study is ready for FB, the IRB analyst will add the study to the next available meeting
agenda.
• If the study team adds a site not previously listed in the IDE or IND protocol, they should submit a
new modification to update the protocol with the new site information (see ‘Modification (MOD)
submission screen process for S-I studies’ for more details).
• If the non-Emory sites include an international site, the Emory sponsor should provide:
o Analysis from Emory’s General Counsel review.

Table of Contents
Page 117 of 273
o Approval letters from the international site IRB/EC review.
o Include the use of a CRO with expertise in the rules and laws of the international site in the
protocol’s site monitoring plan section
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
6/21/2024
Updated throughout to reflect current process and considerations.
8/15/2024
Removes reference to ODP review and updated required training

Table of Contents
Page 118 of 273
SOP Title:
CR screen process for S-I studies
SOP
S-I Submission Management
Established:
7/26/2012
Last Revision:
8/15/2024
PURPOSE
The purpose of this document is to describe the review process of continuing review (CR) submissions by
the IRB analyst when the study involves an Emory Sponsor or an Emory Sponsor-Investigator (S-I)
holding an IDE for a significant risk device or IND.
RESPONSIBILITIES
• IRB analyst – verifies that the information in eIRB is complete, requests the S-I CR checklist, and a
copy of the most recent IND Annual Report or IDE Progress Report.
• Emory investigator holding IND or significant risk IDE: makes sure that the information submitted in
eIRB and on the Emory IND or IDE Responsibilities Checklist is complete and accurate, according to
the study protocol submitted to the FDA.
PROCEDURE FOR CONTINUING REVIEWS
• The IRB analyst will request that the Emory Sponsor/S-I complete the Emory Sponsor
Responsibilities Checklist Continuing Review Update and place it in the submission.
• Once the Emory Sponsor Responsibilities Checklist Continuing Review Update is complete and
information provided confirmed, you can move forward with review.
• If the study is in data analysis only or if the IND is withdrawn (inactive), the Emory Sponsor
Responsibilities Checklist is not required.
• The IRB analyst will ensure that the IND or IDE annual or progress report submission to FDA is
included in the continuing review application under question 7 ‘Attach supporting documents’.
o If the annual report submission is not included in the application, the IRB analyst will determine
from the anniversary date that the IND or IDE went into effect by reviewing the date of the
‘Study May Proceed’ correspondence.
o If the date occurs in the future, the IRB analyst will remind the team to submit the annual
report at the next continuing review.
o If the anniversary date has already passed, the IRB analyst will request a copy of the annual
report submission.
o If the annual report submission is not provided and the annual reporting date has passed (within
60 days of the IND Effective date) the IRB analyst will notify the team to submit an RNI.
o The continuing review can still be assigned to a Full Board committee meeting without the IND
or IDE annual report; however, this might be deemed a pending issue by the committee.
• When the continuing review is ready for Full Board review, the IRB analyst will add it to a meeting
agenda and note the status of the IND/IDE annual report submission.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
6/21/2024
Updated throughout to reflect current process and considerations.


Table of Contents
Page 120 of 273
SOP Title:
Modification (MOD) submission screen process for S-I studies
SOP
S-I Submission Management
Established:
7/26/2012
Last Revision:
8/15/2024
PURPOSE
The purpose of this document is to describe the review process of modification (MOD) submissions by
the IRB analyst when the study involves an Emory Sponsor or an Emory Sponsor-Investigator (S-I)
holding an IDE for a significant risk device or IND.
RESPONSIBILITIES
• IRB analyst – verifies the changes in the submission, and, if applicable, requests the S-I to comment
on whether FDA IND Amendment filing is necessary.
• Emory investigator holding IND or significant risk IDE: makes sure that the information submitted in
eIRB is complete and accurate, according to the study protocol submitted to the FDA.
PROCEDURE FOR MODIFICATIONS TO ADD NON-EMORY STUDY SITES, CHANGE SPONSOR OR
INVESTIGATOR, AND/OR MODIFY THE PROTOCOL
Note for any change to the protocol/IB/IC: if there are any delays in the submission of these documents
(check cover letter or dates on the documents), please check with Team Q as the study team may require
the submission of an RNI.
CHANGE SPONSOR
• The IRB analyst will confirm that the Sponsor submitted an IND amendment or IDE supplement to
the FDA to change sponsor. If the FDA issues correspondence for the transfer of sponsor this should
be provided as well.
• The IRB analyst will verify that the FDA amendment/supplement was submitted and that the
protocol and ICF have been changed accordingly.
CHANGE OF PRINCIPAL INVESTIGATOR
If the study changes the study PI, without changing the Emory Sponsor, the Emory Sponsor is required to
submit an IND amendment or IDE supplement to the FDA. The change must be submitted by the current
PI or Department Approval is required (can be in the form of an email accepting the change for
department leadership). PI Changes require ODP ancillary review before final approval can be issued.
The study analyst will verify that the FDA amendment/supplement was submitted and that the protocol
and ICF have been changed accordingly.
TEMPORARY CHANGE OF PI OR S-I STATUS
For studies that are not in data analysis only: If there is a situation that requires the PI or S-I to
temporarily relinquish their role on the study due to a leave of absence (e.g. maternity leave, sabbatical)
for a period of ≥3 months, a new faculty member should be appointed to fulfill this role in their absence.
A modification to update the record in eIRB, protocol, and consent form is required to be submitted in
this scenario.
S-I MOVES TO ADJUNCT STATUS

Table of Contents
Page 121 of 273
In situations where an Emory S-I leaves the institution remains the S-I and maintains an adjunct status
role, the study will no longer be considered a S-I study when the following criteria are met:
- The faculty member has a primary affiliation with another institution.
- A different faculty member has been appointed as the Emory PI.
CHANGES THAT MODIFY THE PROTOCOL
If the Emory Sponsor/S-I holding an IND or significant risk IDE is making modifications to the protocol
required to be submitted to FDA as defined by 21 CFR 312.30 / 812.35
, the Emory Sponsor/S-I holding
an IND or significant risk IDE will need to either (1) include documentation that they have submitted the
change to the FDA, (2) add a date for when they will submit to the FDA, or (3) confirm the changes do
not require FDA submission. If the FDA submission date is a future date, the study team is also
responsible for logging a comment in the study, confirming that they have notified the FDA of the
change; this will not be a pending issue for the review and approval of an amendment. The following
language should be included in the approval of the amendment:
“The protocol amendment cannot be implemented until the FDA has been notified of the protocol
amendment. If FDA responds with any concerns with the protocol amendment the IRB should be notified
via a new modification.”
If there is any indication that the FDA has concerns with the proposed amendment the IRB should be
provided with these details.
Studies where the Emory Sponsor/S-I holds an IND
Substantial modifications to the protocol must be submitted to the FDA and IRB. Substantial
modifications include any change in a Phase 1 protocol that significantly affects the safety of subjects or
any change in Phase 2 or 3 protocol that significantly affects the safety of subjects, the scope of the
investigation, or the scientific quality of the study. If the IRB analyst is unsure whether the change would
be considered substantial requiring FDA notification the team should be asked to clarify their plans for
notifying FDA and why the change does or does not meet the definition of substantial. Examples of
changes requiring an amendment under this paragraph include:
• Any increase in drug dosage or duration of exposure of individual subjects to the drug beyond that
in the current protocol, or any significant increase in the number of subjects under study.
• Any significant change in the design of a protocol (such as the addition or dropping of a control
group).
• (The addition of a new test or procedure that is intended to improve monitoring for, or reduce the
risk of, a side effect or adverse event; or the dropping of a test intended to monitor safety.
Studies where the Emory Sponsor/S-I holds a significant risk IDE
A sponsor must obtain (FDA and IRB) approval before implementing a change to an investigational plan
unless the change does not affect:
• The validity of the data or information resulting from the completion of the approved protocol, or
the relationship of likely patient risk to benefit relied upon to approve the protocol;
• The scientific soundness of the investigational plan; or
• The rights, safety, or welfare of the human subjects involved in the investigation.

Table of Contents
Page 122 of 273
CHANGES ADDING SITES TO THE PROTOCOL
If the study team adds a site (not previously listed) to the IDE or IND protocol, the study team should
submit a new modification to update the protocol. For IDE studies IRB approval is required before FDA
will approve the addition of sites.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
6/21/2024
Updated throughout to reflect current process and considerations.
8/15/2024
Removed reference to ODP

Table of Contents
Page 123 of 273
SOP Title:
Closeout submission screen process for S-I studies
SOP
S-I Submission Management
Established:
7/26/2012
Last Revision:
8/15/2024
PURPOSE
The purpose of this document is to describe the review process of closeout submissions by the IRB
analyst when the study involves an Emory Sponsor or an Emory Sponsor-Investigator (S-I) holding an IDE
for a significant risk device or IND.
RESPONSIBILITIES
• IRB analyst – verifies that the information in eIRB is complete, ensures proper submission of
annual/final reports for IDEs.
• Emory investigator holding IND or significant risk IDE: submits information to IRB as required.
PROCEDURE
Studies where the Emory Sponsor/S-I holds a significant risk IDE
• Once a close-out request is submitted, the IRB analyst will ensure that the Emory Sponsor/S-I
holding a significant risk IDE provides: 1) a copy of the final IDE report (which is due to the IRB & FDA
within 6 months after the completion or termination of the investigation) and 2) confirmation that
they have submitted the report to the FDA.
• If a final IDE report is not available, then the study must be kept open until the step above is
complete.
• If the study approval lapses, even if a close-out is submitted before expiration, the study will be
considered to have lapsed, and an RNI will also be required for completion of the close-out.
• Once all supporting documentation is submitted (final FDA report, RNI if necessary), then the study
can be closed-out using normal close-out procedures (see Close-out Processes
)
Studies where the Emory Sponsor/S-I holds an IND
Once a close-out is submitted, the IRB analyst will ensure that the Emory Sponsor/S-I holding an IND
provided a timely annual report to the FDA (if one is required) or submitted an IND withdrawal.


Table of Contents
Page 125 of 273
SOP Title:
S-I, Single IRB Study Screening Process
SOP
S-I Submission Management
Established:
1/20/2021
Last Revision:
8/15/2024
PURPOSE
To describe the additional steps to review an S-I study when Emory is serving as the sIRB for external
sites.
SCOPE
S-I studies where Emory is serving as the sIRB for external sites.
RESPONSIBILITIES
• IRB reliance specialist – Responsible for the initial screen of the study, and subsequent follow-on
submissions and for completing the reliance process and processing the addition of external sites in
the smart form. This includes confirmation of required S-I training.
PROCEDURE
Initial Study Review
• The study will be assigned to a reliance specialist based on assignment criteria.
• The reliance specialist will confirm that the use of a single IRB is required, and that the Emory IRB
agreed to serve in this role.
• If Emory is not willing to serve as the sIRB for the study, the reliance team will provide guidance
on how to proceed and the study will be reassigned to a S-I specialist.
• If use of a single IRB is not required, Emory may choose to review for the Emory site and the
study will be reassigned to a S-I specialist.
• If use of a single IRB is required, Emory may rely on an external IRB.
• The reliance specialist will follow the SOP entitled “New study screen process for S-I studies” to
screen the Emory submission and also the SOP entitled, “Processing New Studies When Emory is the
Single IRB of Record (sIRB) for a MultiSite Study” to set up the study as a sIRB study.
• When the study is approved, the reliance specialist will include this statement in the approval letter
“The Emory IRB has approved the study-wide protocol and master consent form(s) and has agreed
to serve as the IRB of record for this multi-site study. External sites have not yet been approved and
may not begin conducting research activities until official site-specific approval has been received.”
Adding Participating Sites (pSites)
• After the study has been approved, the reliance specialist will follow the SOP titled “Study
Processing When Emory is the Single IRB of Record (sIRB) for a Multi-Site Study.”
• The reliance specialist will assign the pSites to the Reliance Team Lead for review.

Table of Contents
Page 126 of 273
• The Reliance Team Lead will approve the pSites or request clarifications.
• The reliance specialist will process the approvals for the sites.
Modifications and Continuing review
• The reliance specialist will follow the SOP entitled “CR screen process for S-I studies”.
Study Closeout
• The reliance specialist will follow the SOP entitled “Closing Out Multi-Site Studies” to close pSites
and the SOP entitled “Closeout submission screen process for S-I studies.”.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/30/2021
Updated process
6/21/2024
Updated throughout to reflect current process and considerations.
8/15/2024
Removed refences to ODP process

Table of Contents
Page 127 of 273
STUDY MANAGEMENT
SOP Title:
Not Research/ Not-Human-Subjects/Not-Human-Subjects-Research/Not-Engaged
SOP Category:
Study Management
Established:
6/28/2013
Last Revision:
04/22/2022
PURPOSE
Outline the necessary steps for processing a determination of not research (NR), not-human-subjects
(NHS), not-human-subjects research (NHSR), or not-engaged. Determinations are made in response to:
• Requests for a determination via MS Form
NON-HUMAN SUBJECTS RESEARCH
DETERMINATION FORM
• Requests for a determination via email (typically after using the form on the website but have
been instructed to contact the IRB)
• eIRB submissions which upon initial review are determined NR/NHSR/NE
SCOPE
This applies to all IRB staff, particularly those who manage listserv and analysts who conduct initial
review of new eIRB submissions.
RESPONSIBILITIES
• Listserv Monitors: Assist investigators to confirm whether IRB review is required or not based on
the responses entered into the MS Form, NON-HUMAN SUBJECTS RESEARCH DETERMINATION
FORM
• IRB Analysts: Conduct initial review of eIRB submissions and determine if the project is
NR/NHS/NHSR. Receive NHSR requests via email when determination form refers requestor to IRB,
and determine whether the project is NR/NHS/NHSR/NE, or if it requires a formal submission in
eIRB. Compose NR/NHS/NHSR/NE letters if required and:
o Send to PI via logged comment, then withdraw the study (if received via eIRB submission).
o Send to PI via email (if received via listserv).
PROCEDURES
REVIEW OF NR/NHSR/NE DETERMINATIONS
NR/NHSR/NE determinations are typically made in response to either MS Forms NON-HUMAN SUBJECTS
RESEARCH DETERMINATION FORM requests, email requests after the determination form refers
requestor to IRB, or upon initial review of eIRB submission. These scenarios are separated into respective
sections below:
Email Requests for NR/NHSR/NE Determinations – No prior MS Forms submission

Table of Contents
Page 128 of 273
If not clearly HSR, request that the person use the MS Forms NON-HUMAN SUBJECTS RESEARCH
DETERMINATION FORM tool first and come back if the tool determines IRB consultation is needed. If
requestor has been told a formal letter is required, skip to “Email Requests for Formal NR/NHSR/NE
Determinations – MS Forms not accepted by third party,” section below.
Email Requests for NR/NHSR/NE Determinations – Requestor has already used MS Forms
1. If the investigator has not provided any attachments that may include their NON-HUMAN SUBJECTS
RESEARCH DETERMINATION FORM request responses, you will first need to locate the investigator’s
NON-HUMAN SUBJECTS RESEARCH DETERMINATION FORM responses.
a. Ensure that you are logged into Emory Office 365
b. Click on the “waffle” in the upper left-hand corner of the display and navigate to the Office
365 App Forms
c. When you first navigate to Forms, you may not see any Forms to choose from. Scroll down
until you see “My Groups” and click on IRB-Staff. The view may look like this:
d. Click on NON-HUMAN SUBJECTS RESEARCH DETERMINATION FORM – there will be two tabs,
Questions and Responses, click on Responses. Click on the “Open in Excel” icon.

Table of Contents
Page 129 of 273
e. Search the Responses Excel spreadsheet for the investigator’s name or email address
f. If you want to review their actual responses in the Form layout, you can click on the blue
“View results” button
g. If you have already found the Respondent ID number from the spreadsheet you can enter it
here:

Table of Contents
Page 130 of 273
h. Review the investigator’s responses to determine which item on the form recommended that
they submit their project proposal to the IRB for review and confirmation that their project does
not constitute human subjects research at Emory. You will also need to read the project
proposal.
i. Either ask the study team to correct the MS Form if they made an error and it should have
resulted in a “no IRB required” determination or confirm that they need to submit in eIRB.
Email Requests for Formal NR/NHSR/NE Determinations – MS Forms not accepted by third party
1. Take a preliminary review of the information submitted about the project; determine whether
additional information is required
a. The NON-HUMAN SUBJECTS RESEARCH DETERMINATION FORM requires most basic information
to be included (emails directly to the listserv may be lacking)
b. Typically it is necessary to have a copy of the protocol summary along with information about
objectives, procedures, funding and data source
2. If the project involves the VA in any way (study site, affiliation of researchers, data source, etc.),
send the determination request to the Emory/VA liaison. Copy the person who made the request
when you forward the email to the liaison to let the person know that the request has been received
and it is being rerouted.
3. After receiving all the necessary information, consider whether the project qualifies as human
subjects research
a. Is it a “systematic investigation designed to contribute to generalizable knowledge?” Does it
involve human subjects with whom an investigator has interaction or intervention, or about
whom an investigator receives identifiable private information? (See 45 CFR 46.101)
1) Remember that the FDA has a different interpretation of human subjects; even if using only
de-identified samples, clinical investigations that support applications for research or
marketing permits for products regulated by the FDA require IRB review.
b. There are several resources to aid your determination; refer to the guidance folder and consult
with colleagues for assistance.
4. Reply by email to the investigator with an official determination
a. The email should state that the IRB has reviewed the provided information
and determined that the project does not require IRB review because it (a)
does not meet the definition of “research” or (b) does not involve “human
subjects” or (c) Emory is not “engaged” in the human subjects research.
(Refer to the letter template for guidance on the language to include in the
email: “H:\General\Admin IRB Documents\Checklists, Forms, and
Templates\Letter templates and suggested language\NHS NR Letter
Template.doc” and “\Not Engaged Template Letter”)
b. The more specific the better, remember that we may need to refer back to
this determination in the event that a concern arises later. Cite specific
regulations if possible, and explain any possible points of confusion. For
example, “…not research because this is a quality improvement/case
report/educational initiative designed to…”
c. At the end, explain that the email is sufficient documentation of the IRB’s
determination but if a more formal letter of determination is necessary in
the future please let us know.
5. Document the determination on the H: Drive

Table of Contents
Page 131 of 273
a. Create a new folder under H:\General\NR-NHS Letters\[Last Name, First]
b. Add to the new folder:
a. Submitted documents (e.g. protocol, survey, etc.);
b. PDF of all email correspondence;
c. The formal letter of determination, if drafted.
c. Remember: this documentation is the only proof that the IRB will have in
the future. If, for example, an academic journal requests our reasoning for
not requiring IRB oversight, we need to have this documentation. It’s also
necessary for internal record reviews, and possibly external audits.
eIRB Submissions Determined NR/NHSR or “Not Engaged”
1. Review the eIRB submission and determine whether the project constitutes human
subjects research, and if so, is the Emory team “engaged” in the research
a. Is it a “systematic investigation designed to contribute to generalizable
knowledge?” Does it involve human subjects with whom an investigator
has interaction or intervention, or about whom an investigator receives
identifiable private information? If a multisite study does involve HSR, is
Emory actually doing any of it (i.e. are we “engaged”)? (See 45 CFR 46.101)
1) Remember that the FDA has a different interpretation of human
subjects; even if using only de-identified samples, clinical
investigations that support applications for research or marketing
permits for products regulated by the FDA require IRB review.
2) Also remember that if Emory is the prime awardee of a federal
grant where the grant proposal was submitted as human subjects
research, we are still considered “engaged” in the research, even if
we’re farming out the human subjects research activities.
b. There are several resources to aid your determination; refer to the
guidance folder and consult with colleagues for assistance.
2. If determined NR/NHSR or “Not Engaged,” submit the pre-review, documenting any
regulatory oversight that applies. A pre-review checklist is not required.
3. Assign yourself as a designated reviewer and conduct the review, noting that the
project is not research with human subjects.
4. Finalize only the protocol so that it is referenced in the letter that’s generated.
5. Click “Prepare Letter” to generate the appropriate letter. Draft the letter and upload
it.
o Be sure to include in the letter a brief rationale for your determination,
making it clear whether the work is “not research,”“not human subjects,” or
that Emory is “not engaged,” and why.
6. Click “ok.”
7. Click “Send Letter” to send the letter to the team. The study should not have an
expiration date since it was not “approved.”
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
04/22/2022
Large overhaul of SOP: Addition of instructions for how to handle when the NON-HUMAN SUBJECTS
RESEARCH DETERMINATION FORM responses indicate that the investigator should consult the IRB via
emailing the IRB listserv.Also included “Not Engaged” determinations.

Table of Contents
Page 132 of 273
MODIFICATIONS
SOP Title: Adding/Removing Study Personnel
SOP Category:
Modifications
Established:
4/11/2014
Last Revision:
8/15/2024
PURPOSE
This SOP describes the process for IRB staff to process Modifications that include only staff changes.
SCOPE
The SOP applies to
all approved, non-expired studies.
Note: If the personnel change is for a study with AVAHCS (the Atlanta VA) as a Research Location, only
the VA liaison or their backup can process the modification as there are additional considerations. See
the last step.
PROCEDURE
1. Click the “My Inbox” tab if not already there.
2. Click on the “Personnel Changes” tab.
3. Click “Date Modified” once or twice to sort that column by date order, newest to oldest.
4. For each one showing you as the assigned Coordinator, click the link to the Modification under the
“Name” column. Right click and select “Open in new tab” if desired.
5. Click on “View Modification/CR” on the left menu.
6. Confirm the modification summary states the names of the people being added and removed. Click
“Compare” on the top left and review the changes that are indicated by a pencil icon to ensure the
changes have been made to the smart form.

Table of Contents
Page 133 of 273
7. For all study staff being added to the smart form as “Local Study Team Members,” confirm the
person is affiliated with Emory by entering the person’s name in the Emory Online Directory.
• If the person’s name does not appear in the Directory, log a comment to the study team stating
you are unable to locate them in the directory and asking for clarification about their affiliation
with Emory or CHOA.
CHOA study personnel should be listed in an excel spreadsheet or external team
member list under the External Team Member section and they are to upload the
pdf CITI training certificate (not transcript) in that same section of the smart form.
If the study team confirms they are adding someone who is an external study team
member (other than CHOA), please direct them to the guidance posted on our
website under Collaborative Research.
If the study team is making changes to local study team members as well, tell them
to remove the external study team members and submit with a future mod
following the guidance on our website.
Note: If the modification includes a change in the PI, this requires a Modification with “Other parts of
the study” selected as the scope and requires changes to documents.

Table of Contents
Page 134 of 273
8. Click “View CITI training” on the left menu (see below) and verify that and “Local Study Team
Members” being added have the required CITI training for the study. Review the
“Training
verification” SOP for more information.
• External Study Team members who are not affiliated with an institution must complete Emory’s
CITI training and upload their CITI certificates (not transcripts) into the External Study Team
section of the smartform. (External collaborators who are affiliated with an institution do not
need to upload their CITI certificates. Their local IRB will document they have current CITI
training via the local context review form.)
9. If any study staff are missing required CITI training, log a comment to the study team letting them
know which study team members need to renew their CITI training and that if they do not populate
in the View CITI Training feed, they can upload the certificates (not transcripts) with a logged
comment.

Table of Contents
Page 135 of 273
10. After confirming valid training for all study staff being added, click “Submit pre-review” in the left
menu (see activity right under “Printer Version”).
11. A new window will open. Scroll to question 8.0 and click “Yes”. Do not edit any other selections.
12. Click “Assign Designated Reviewer” in the left menu (see below).

Table of Contents
Page 136 of 273
13. Select yourself as the designated reviewer from the list (click the upside-down little arrow on the
box in the first question), then click “OK” at bottom-right of the popup window.
Click “Submit Designated Review” on the left menu.
14. Select “Approved” as the determination.
15. Select “expedited” as the review level.
16. Select “minor modification” as the category.

Table of Contents
Page 137 of 273
17. If you are asked to provide reasoning why a study needs to come back for continuing review, write
“This is just a study staff change.”
18. Scroll to question 9 and check the box if no conflicting interest. Then click “Yes” for question 10.
19. Click “Prepare Letter” on left menu.
20. In the new window shown below, select the “Acknowledgment of Personnel Update” template and
click the “Generate” button, then click “OK.”
21. Click “Send Letter” on left menu.

Table of Contents
Page 138 of 273
22. In the window that opens, click the link to the letter to make sure it is correct. Click OK to send the
letter.
23. The submission will be complete after this step.
24. If the study is a VA study, add the effective date of the study staff addition/change/ removal (the
date the VA Staff Changes Request form was approved by the VA WOC coordinator) to the body of
the approval letter. Note that no VA staff additions/changes/removals may be processed without
the approved VA Staff Changes Request form. The VA study records are updated before the Emory
IRB study records and reflect the VA approval of the staff update(s).
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/13/2015
Added purpose and scope and update of the information
6/22/2016
Added guidance for incomplete requests
5/17/2018
Added information to respond to ineligible requests, replaced SOM contact name for volunteer
verification and added VA information
2/11/2020
Overhaul of this SOP due to change of electronic system
2/26/2020
Added more detail about how to look for changes in the submission, and added graphics to identify
MODS that should not be approved by the student workers
3/4/2020
Adding instructions in case the system ask for a justification of why a CR needs to take place again
5/18/2020
CHOA personal can be added under External personnel
11/3/2020
Adding information for VA studies
7/5/2022
Many minor edits for clarity and added step to confirm study staff being added are in Directory


Table of Contents
Page 140 of 273
SOP Title: Modifications- Processing from Preliminary Analysis through Approval
SOP Category:
Study Management
Established:
8/9/2013
Last Revision:
07/6/2023
PURPOSE
The purpose of this SOP is to outline the step-by-step procedures for processing a Modification from
submission to approval.
SCOPE
This SOP applies to Modifications that need both expedited and full board reviews. If the modification is
for staff change only, ignore as the dedicated staff members these modifications will take ownership of
them. This SOP only applies to the modification process. Please see CR SOP for details on handling
renewal submissions.
DEFINITION
• Modification: Any change to a current study that would require review from the IRB before
implementation.
• Ancillary Committee: Refers to outside committees including Biosafety, Radiation Safety (RSC),
PRMC etc., that would need to review a Modification based on any additional procedures being
added that would trigger their departments review.
RESPONSIBILITIES
• IRB Analyst: Take the Modification through the entire process from submission to approval
PROCEDURES
Analysis of Modification Application
Note: Find all the checklists on this SOP in this folder H:\General\Admin IRB Documents\Checklists-
Staff, Forms, and Templates\1. Staff Screening Checklists and Tip Sheets.
• Have the MOD screening guide accessible while analyzing the Mod application H:\General\Admin
IRB Documents\Checklists, Forms, and Templates. For more information about pre-review and
ancillary review information, review our SOP
.
• Lapsed studies: add to the modification approval letter that no research activities can take place
unless the study is re-approved after the approval of a continuing review application.
• If the Mod includes a 483 report after an FDA inspection to the study or sponsor, ask the study to
remove this report and to submit via RE.
• If this Mod adds a drug under REMS or a drug considered a controlled substance;

Table of Contents
Page 141 of 273
o Studies using controlled substances drugs should fill out the Investigator Checklist for the Use of
Schedule I Controlled Substances and email to Margaret Huber at mhuber@emory.edu. Make
sure you communicate with Margaret about next steps.
o REMS process has been followed, per our SOP entitled: “REMS study review”. Please review
REMS SOP and talk to a TL or IRB Director about following steps
o If adding a new funding source, please reference Guidance to connect Grants and IRB Approved
Protocols. If new DoD funding, see study checklist. The amendment screening guide includes
additional considerations around new funding sources.
Verify that the proposed modifications have been made in the revised eIRB smartform and Mod
application. For guidance on viewing changes please see the Huron Staff Quick Reference
Document.
If changes are needed, click the “Request Pre-Review Clarification” and inform the study team of the
requested changes.
• The state of the Mod will change to “Clarification Requested (Pre-Review)”
• Follow up with the study team if a response is not received in a timely fashion. If a submission is in
requested changes for more than 30 days with no answer from the study team, the analyst should
withdraw it. The analyst needs to document their efforts in contacting the study team and email
them before withdrawing the study.
• The study team will need to hit the “submit response” button to return the Mod back to the IRB to
be processed. Sometimes the study team will make changes without submitting the study back. Be
sure to occasionally review the “In Process” tab for logged comments to catch these Mods.
If no further changes are needed, determine the appropriate routing of the review (i.e expedited or
full board). Please also complete the Pre-Review Note: Do not modify selections under “Regulatory
oversight” and “Special determinations and waivers” sections unless the Modification impacts the
applicable area. Reference the following SOPs to determine the appropriate route and reviewer of the
Mod:
• Determinations and Reviews by IRB Staff:
for minor administrative changes that can be approved by
the analyst assigned to review the Mod
• Categories of research reviewable by IRB staff as IRB designated members: for selected Mod that
could be reviewed by Associate or Assistant Directors or other Sr. RPAs.
• Modifications Indicating Increased Risk: for a Mod that may require Full Board review
For guidance on assigning for designated review, please see the Huron Staff Quick Reference
Some considerations when assigning reviewers:
• For expedited studies: Assign expedited reviews to the reviewer whose specialty is most closely
related to the study. For full board reviews assign to the committee who has a representative who
has the needed specialty.
• For exempt studies: In situations where IRB staff can complete initial review for an exempt study,
analysts can self-review Modification submissions. Modifications for studies that underwent limited
IRB review initially (reviewed by a staff designated reviewer) must also be sent to a staff designated
reviewer for a determination that the study’s exempt status has not changed.

Table of Contents
Page 142 of 273
• Under the Comments section, include a brief description of the proposed Mod when assigning the
Mod for review. The description should allow a reviewer to quickly ascertain the contents of the
Mod.
• Press “OK”
• The current state of the Mod should change from “Pre-Review” to “Non-Committee Review.”
• At this point you wait for the expedited reviewer to complete their review
After the review is submitted the state will change to “Post-Review”. Go to the “Reviews” tab to find
the reviewer's determination.
• If changes are requested, select “Prepare Letter” and choose the template for “Modifications
Required to Secure Approval”
Once your letter has been created, click “send a letter” and wait for changes to be submitted
back from the team.
Screen the changes. If the changes seem complete, do not click on “review required
modifications”. Instead, click on “assign designated reviewer”. When sending the submission to
the reviewer, make sure that your comment explains this is a contingency review. After the
designated reviewer has submitted their review, ensure that the approval/expiration dates,
especially after a pending review, are correct. You can submit a review yourself to correct the
issue, adding a comment of why you are resubmitting. If unsure, ask one of the TLs.
If you are ready to finalize approval, first review the expiration date in the designated review space.
Ensure that the expiration date is the overall study expiration date.
o If the expiration date is incorrect, click on “submit designated review” and correct it.
• After this check, click “Finalize Documents” on the study workspace. Select only the new or revised
documents to change to PDF and watermark. Click OK. The Documents tab on the study workspace
will include links to the final versions of the documents.
If no changes are requested, select “Prepare Letter” and choose the appropriate approval template
from the drop-down selections.
Tips for drafting letters:
• Choose an appropriate letter template and generate it from the dropdown menu. Download the
generated letter by selecting “Download Copy” and make the following edits:
• Remember to include the PI’s post-nominal (degree – MD, PhD, MPH, etc.)
• Delete the template letter language that does not apply to the current MOD and remove rows that
state “No Items to Display.”
• Edit the “ENTER NAME OF LETTER SIGNATORY” and “title’ fields to add your name and title. Save a
copy of the letter on your desktop and “upload revision"

Table of Contents
Page 143 of 273
• Click “Send Letter”
• MOD/CR (see CR SOP for details on processing the CR portion of the submission). Reference the
Huron Staff Quick Reference
for details about handling this type of submission.
a. Ensure that all documents were finalized appropriately by opening them from the
“Documents” tab.
Full Committee Modifications:
• To start, log a private comment indicating why the modification requires Committee review.
• See the Huron Staff Quick Reference
for details on assigning for Committee review.
• Click on “Edit pre-review”, and add the following under question 6 (Notes):
o Add if the study is currently enrolling subjects (especially if the study team did not
answer that question in the modification submission)
o If adding study personnel, if the CITI is not up to date (this will be a pending item
otherwise)
o If this is a Sponsor-Investigator study:
If this Mod is adding new sites (Y/N)
If yes, the responsibility form should be included in the submission with an S-I
consult approval (This is required before the Mod can be approved)
o If this is a Risk Evaluation and Mitigation Strategy (REMS) (Y/N)
o If yes, has a REMS consult approval submitted? (This is required before the Mod can be
approved)
Steps to Check the Full Board Committee’s Decisions Reviewer’s Review
Once the meeting pod has reconciled the meeting notes and omnibus forms and releases the agenda for
letter writing via email, review the applicable forms to obtain committee decision.
• Follow steps outlined above and in the Huron Staff Quick Reference
for further processing for either
“modifications required” or approval.
• If modification is deferred, once the team submits changes check that deferred issues have been
addressed, schedule the Deferred modification to be reviewed by the same committee that
conducted the initial review of the modification application (i.e. if CMTE B1 reviewed the
modification in the January meeting and deferred it, send to the next available meeting for CMTE
B1).
• After the meeting, if the Deferred issues are approved, follow the steps for processing approval.
• If a modification is pending approval, once the team submits the changes:
o Email the study link to the designated reviewer. Ask them to review the changes but to not
make any changes in the submission. They may log a private comment or email you back. If the

Table of Contents
Page 144 of 273
member does not log a comment, make sure you log a private comment with their email
response.
o Click on “Review Required Modifications” The “approval date” would be the date of the
meeting. The “effective date” is the date the vicechair confirmed the changes were adequate
and the study can receive final approval.
o You can now proceed with generating a new approval letter and sending it.
• Follow up with the study team if a response is not received in a timely fashion. If a submission is in
requested changes for more than 60 days with no answer from the study team, the analyst should
withdraw it. The analyst needs to document their efforts in contacting the study team and email
them before withdrawing the study.


Table of Contents
Page 146 of 273
SOP Title:
Modifications: Mods Indicating Increased Risk
SOP Category:
Study Management
Established:
7/18/2012
Last Revision:
8/30/2021
PURPOSE
To explain the process of reviewing Modifications (Mod) that indicate an increase in risk, and to describe
when a reportable new information (RNI) submission may be necessary.
SCOPE
The SOP applies to studies approved by the Emory IRB.
RESPONSIBILITIES
• IRB Analyst - will review the submission and assess if the Mod may need a RNI submission.
• IRB Team Q member – will assist analyst in determining if an Mod needs a concurrent RNI
submission.
PROCEDURE
1. Determine if an RNI should be submitted along with the Modification:
a. If this Mod information comes from an analysis of aggregated data by the sponsor (action
letter, post-marketing data analysis, etc.) and if it appears to be unrelated to a specific
adverse event, then no reportable new information submission is required.
i. For example: the Mod states that, after reviewing data from all sites, they have discovered
an increased frequency of event X, and they have decided to modify the protocol, ICF or IB.
Such sponsor-prompted Mods do NOT require a concurrent RE. This is because the sponsor
has done a review of these aggregated data and the IRB would have no additional
information with which to do a better review, or call something an UP. The IRB’s task now is
to review the proposed changes to protocol, IB and especially the ICF. This process can be
completed by the Full Board, without the need of a reportable new information submission.
b. If the Mod information includes a safety memo about an event involving an individual
subject or subjects, a reportable new information submission may be required.
i. For example, the Mod may state that X number of subjects experienced event X, detailing
each clinical case in the report, and that is why the sponsor thinks the matter is unexpected,
serious and will make changes to the protocol, ICF or IB. The study analyst should alert
Team Q about the Mod. Team Q will verify if a RNI is required or not. If Team Q received a
RNI that requires the submission of an Mod (if not already submitted), Team Q will log a
comment into the study history to alert the study analyst.
2. Determine the mode of review for this Mod: When receiving a new risk via a Mod, check
this
document to verify if the information in the Mod was previously reviewed at FB and can go
expedited in subsequent studies. If not, the new risk information should go to FB EXCEPT when it
falls under 3(b) below.
*Note: If there are changes to the Investigator’s Brochure (IB) without ICF changes, then ask study team
when changes to ICF are expected. If the study team said the changes to ICF are forthcoming, send to FB
with a note explaining the study team has not provided ICF changes. Note in the omnibus form under
“pending” section in Mod.

Table of Contents
Page 147 of 273
3. Handling of Modification with respect to Reportable new information submission (when required)
a. If a reportable new information submission HAS been submitted and it HAS NOT already
been reviewed by CoRe or Full committee (i.e., a determination of UP or not an UP has not
been made), then analyst should contact the case manager for the reportable new
information submission for more information. Case manager should also log a comment in
Mod and communicate with study owner about RNI disposition.
b. If the CoRe determines that the reportable new information submission is not a potential
UP, and determines that there is no change to the overall risk/benefit ratio of the research
study, then the Modification may go to expedited review.
c. If the CoRe determines that the reportable new information submission is a potential UP
that represents an immediate safety concern, then it must be reviewed at the next full
board meeting. The IRB analyst should add the Modification to the same meeting’s agenda
where the reportable new information submission will be reviewed, if possible. In general,
Modifications and REs with new safety information should be reviewed in the next available
meeting. Please, discuss this with the Q Team and meeting facilitators.
d. If the CoRe determines that the reportable new information submission is a potential UP
that does not represent an immediate safety concern, then it will be reviewed at the next
Committee Q meeting . The Modification can also be reviewed at the Committee Q meeting
if the only changes relate to the reportable new information submission and/or other
changes do not need to be reviewed sooner (discuss with your Associate or Assistant
Director or Director if not sure).
i. Let the Team Q case manager and Q meeting facilitator know that the Modification
will be reviewed at Committee Q so materials can be prepared. After the Team Q
Meeting, the case manager will log a comment in the Modification history detailing
if the Modification was approved as it is or if it has pending issues.
ii. If the Modification was approved, the analyst should assign the review to
him/herself for completion. This does not mean the analyst is making a
determination; the Modification was approved at the Committee Q. The analyst is
simply completing the review to move the Modification to the draft letter state.
iii. If the Modification has pending issues, the Modification should be assigned to the Q
chair (Dr. Stein) for review and final approval.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/30/2021
Removed reference to old location for tracking new risks and archived log of updates older than 1
year

Table of Contents
Page 148 of 273
NEW STUDIES
SOP Title: Pre-Review options and Ancillary Review Information
SOP Category:
Study Management
Established:
02/24/2020
Last Revision:
08/15/2024
PURPOSE
To clarify the appropriate selections in the eIRB “Submit Pre-Review” and “Manage Ancillary Reviews”
activities, based on the details of the submission.
SCOPE
The SOP applies to all studies reviewed by the Emory IRB.
RESPONSIBILITIES
• IRB Analyst: Makes the appropriate “Submit Pre-Review” and “Manage Ancillary Reviews”
activity selections in eIRB as part of their comprehensive Pre-Review of study submission
• Analyst Supervisors: Assists IRB Analysts, as needed
PROCEDURE
Submit Pre-Review Activity
Once the IRB Analyst has completed an initial review of an IRB submission in eIRB, select “Submit Pre-
Review” on the left side of the submission page. The Pre-Review activity page will open as a separate
window. Analysts should reference a study’s protocol, consent, and funding sections in the eIRB
submission to complete the Pre-Review activity.
If ongoing updates to the Pre-Review or study submission are needed, select “
For new study submissions: Review and complete all sections of the form as appropriate.
For Modification and/or Continuing Review (CR) submissions: Confirm the selections are accurate and
make updates, as appropriate. Note that the first question, “Common Rule Regulatory Requirements,”
will only be shown in the Pre-Review for a CR submission.
Pre-Review Selections
1. Common rule regulatory requirements
Note: This appears for CR submissions currently under Pre-2018 Requirements, as #1.
• Pre-2018 Requirements: Select if the study is FDA-regulated, DOJ-sponsored, or not migrating
to the revised common rule.
• 2018 Requirements: Select if eligible for transition to the revised common rule and actively
being transitioned with the CR submission.
1. Regulatory oversight: Select all that apply to the study.

Table of Contents
Page 149 of 273
Note: If selection funding-based, select the funder even if the funds come from a “pass through”
source (i.e., If Emory is receiving a sub-award from a university for a federally funded project, select
the primary funding source in this section). For example, if the Emory receives a sub-award, and the
sub-award is paid for with DOD funds, the study is considered DOD funded.
• DOD (Department of Defense): Select if funding is received from the agency.
• DOE (Department of Energy): Select if funding is received from the agency.
• DOJ (Department of Justice): Select if funding is received from the agency.
• ED (Department of Education): Select if funding is received from the agency.
• EPA (Environmental Protection Agency): Select if funding is received from the agency.
• FDA (Food and Drug Administration): Select if one of the following applies:
o Funding is received from the agency
o The research directs the use of a drug, biologic, dietary supplement or medical food, or
any other regulated investigational product, even if FDA-approved
o The research includes a device or medical mobile app/software and evaluates its
safety or effectiveness of the device, even if FDA-approved
o Notes:
If eIRB questions #1 or #2 under Study Scope are marked yes, then selection is
required.
If eIRB question #3 under For Clinical Research/Expanded Access Only is
marked yes, then selection is required. Notify Team Q
prior to completing Pre-
Review to ensure all regulatory requirements are addressed.
• GDPR (General Data Protection Regulation): Select if this study plans to collect data about
people located in the EEA (European Economic Area). See
international study guidance posted in
the H:\Drive.
• HHS (Department of Health and Human Services): Select if funding is received from the agency,
even if through a “pass through” source. This includes the following common funders:
o Note: this is not an exhaustive list of HHS funding sources
o CDC (Centers for Disease Control and Prevention)
o CMS (Centers for Medicare and Medicaid Services)
o NIH (National Institutes of Health)
Note: Many funders with “National” in their title or abbreviated starting with “N” are a
part of NIH. Reference the NIH’s list of institutes and centers
. Centers that do not fall
under this approach include:
CFAR (Center for AIDS Research)
CC (Clinical Center at NIH)
FIC (Fogarty International Center)
• ICH-GCP (International Center for Harmonization of Good Clinical Practice): Select if the study
protocol says ICH-GCP, as adopted by FDA.
o If the protocol indicates ICH-GCP E6, notify the IRB Director. This selection still applies.
• NSF (National Science Foundation): Select if funding is received from the agency
• OCR (Office of Civil Rights): Select if any data comes from the medical record, including when:
o HIPAA applies to the study, regardless of if it applies to the research records
o A HIPAA Waiver is granted, including a Partial HIPAA Waiver where no medical data
will be collected apart from recruitment
o Note: If eIRB questions #1 or #2 under HIPAA Applicability and Waivers Requested are
marked yes or #3 indicates a Partial HIPAA Waiver, then selection is required.

Table of Contents
Page 150 of 273
• VA (Department of Veterans Affairs): Select if research occurs at, or is funded by, the VA
• Other federal agencies: If federally funded and none of the above options apply, select this
option and continue processing the study. Contact the IRB Director to determine what Pre-
Review selections are needed prior to finalizing submission approval.
• Tribal Law: Select if research involves participants from a tribal nation or specifically studies a
tribal nation. Contact the IRB Director prior to completing Pre-Review to ensure all regulatory
requirements are addressed.
• None of the above: Select only if no other options apply.
2. Special determinations and waivers: Select all that apply to the study.
Note: If a worksheet is indicated with the selection, make sure to complete the indicated worksheet
and upload with the Pre-Review Checklist.
• Broad Consent: Do not select this option.
• Institutionally, Emory does not permit broad consent for any study. If a study requests
the use of broad consent, let them know this is not allowed. If the study team pushes
back, contact a director.
• Children: Select if minors are enrolled in the study or the research collects identifiable
information about minors.
• Note: select even if only includes survey or chart reviews
• Complete and upload the Subpart D worksheet
• Children who are wards of the state: Select if enrolled in the study or the research collects
information about children who are wards of the state.
• Note: select even if only includes survey or chart reviews
• Complete and upload the Subpart D worksheet
• Cognitively impaired adults: Select if cognitively impaired adults will be enrolled in the study.
• Complete and upload the Cognitively Impaired Adults Checklist
.
• Neonates of uncertain viability: Select if included in the study.
• Note: do not select if incidental to the study (e.g., chart review)
• Complete the Subpart B worksheet
• Nonsignificant risk device: Select if this determination is requested or approved by a convened
IRB Committee. Note these are also referred to as NSR Devices under Abbreviated IDEs.
• Ensure the Device Checklist is completed.
• Do not select if only IDE Exempt devices are included in the study.
• Non-viable neonates: Select if included in the study.
• Note: do not select if incidental to the study (e.g., chart review)
• Complete the Subpart B worksheet
• Pregnant women (includes all pregnant persons): Select if included in the study.
• Note: do not select if incidental to the study (e.g., chart review or survey only)
• Complete the Subpart B worksheet
• Prisoners: Select if included in the study.
• Note: do not select if incidental to the study (e.g., chart review)
• Complete the Subpart C worksheet
• Students/Employees: Select if the study recruits or specifically targets Emory employees or
students or study team members otherwise employ or oversee study participants.
• For example: The study enrolls unit staff for as healthy controls, or the PI approaches
their lab staff to participate complete the study

Table of Contents
Page 151 of 273
• Note: No checklist is required, unless FERPA also applies to the research.
• Waiver of consent documentation: Select if the study includes any of the following, based on
the study’s HIPAA attestation and waiver document
and eIRB selections:
• Waiver of documentation of consent (also referred to as verbal consent or online
consent), including consent for eligibility screening
• Note: Do not select this option if assent is waived (document in the Subpart D checklist)
or if a complete waiver of consent is granted (select the appropriate options, below).
• Waiver of consent for emergency research: Select if applies to the study (also referred to as
exception from informed consent or EFIC).
• Reach out to an IRB Director or Designee to clarify what further documentation is
needed.
• Complete the mysterious hidden form that only special little analysts can find.
• Waiver of HIPAA authorization: Select if the study includes any of the following, based on the
study’s HIPAA attestation and waiver document
and eIRB selections:
• Partial Waiver of HIPAA Authorization (also referred to has PHW, including if only for
recruitment without later authorization)
• Waiver of Elements of HIPAA Authorization
• Complete Waiver of HIPAA Authorization
• Waiver/alteration of the consent process: Only select if none of the above informed consent
waivers are selected.
• Complete waiver of consent (note: this typically for chart review studies only)
• Waiver of elements of consent (note: this is very, very, very rare)
3. Type of research: Select each that applies to the study, based on the topic of the research.
Note: The selection should coincide with the IRB Committee assigned to review the research.
• Biomedical/clinical: biological function, pathology/disease, clinical care, diagnosis, treatments,
public health, epidemiology, psychiatry, and health services (Note: this includes most medical
record reviews)
• Social/behavioral/educational: individual and social behaviors, including sociology, psychology,
anthropology, ecology, history, communications, and business.
• Other: This option should rarely be selected. Contact the IRB Director or their designee to clarify
the appropriate selection if neither of the above options apply.
4. Additional study features: Select each that applies to the study.
• Clinical Trial: Select if the study meets the NIH’s definition of a clinical trial: a research study in
which one or more human participants are prospectively assigned to one or more interventions
(which may include placebo or other control) to evaluate the effects of those interventions on
health-related biomedical or behavioral outcomes.
• This should be selected if the study team responds “Yes” to #1 under For Clinical
Research/Expanded Access Only.
• Note: If you believe that the selection in the smart form is incorrect,
email Jennifer
Prozonic for guidance.
• Certificate of Confidentiality (COC): Select if the study NIH-funded or has been issued a COC.
• Do not select this option if the COC application or NIH funding is pending.
• See the SOP on COCs
for more information.

Table of Contents
Page 152 of 273
• Collaborative: Select if the research procedures are divided between institutions, even if each
institution’s activities are reviewed separately.
• Deception: Select if the study involves deception or incomplete disclosure.
• Multi-Site Study: Select if Emory is following the same protocol/conducting the same
procedures as other study sites, even if each institution’s activities are reviewed separately.
• Note: These submissions will likely require a supplement to the sponsor protocol.
5. Missing materials: List pending items of the submission here.
6. Notes: Provide a description of the submission details here.
Note: If this is a MOD or CR, it is recommended that you include the submission ID with any notes
(e.g., “CR002”) and maintain any key study details already populated in this Notes section.
7. Add supporting documents: Upload any review checklists here.
8. Are you ready to submit this pre-review? Select “Yes” if you are ready to assign the submission for
IRB Review. Otherwise, select “No” if clarifications are needed in the Pre-Review stage.
Ancillary Reviews
Ancillary Reviews are required assessments and evaluations of IRB protocols conducted by groups
outside of the IRB. Analysts should review study protocol, eIRB submission, and team members to
determine what ancillary reviewers apply to the study and initiate these reviews soon as the required
review is identified.
Note: The IRB will not issue approval to initiate research when a study has not been approved by the
ancillary reviewers.
Setting and Checking Ancillary Reviews in eIRB:
On the study submission page, select “Manage Ancillary Reviews” from the left-side submission menu. A
new window will open with all Ancillary Reviews set for the current study and the current review status
(approved, not approved, or incomplete).
• To add a new Ancillary Review: Select “Add” in the window and complete the questions:
1. Under Organization, search for and select the name of the organization for the ancillary
review that is needed or if selecting department review, select the PI’s department. Use the
% wildcard symbol to assist in the search (e.g., “%CHOA” will show all groups listed with
“CHOA” in their name).
2. Review Type: Select the type of ancillary review (e.g., department review, EHSO Radiation
Safety, etc.)
3. Indicate if the response is required based on the review description
• Select “OK” or “OK and Add Another” once all information is confirmed
• To edit the Ancillary Review Information: Once a review is listed, it cannot be edited. If
incorrect information was entered, delete the listed review and generate a new one, as
instructed above.

Table of Contents
Page 153 of 273
• To upload documentation or mark an Ancillary Review as completed: Select “Update” next to
the review to be edited. You can then update options 4-6 under the review listing:
1. If you have documentation that the review was approved (e.g., a letter from the reviewing
group), update this selection to “Yes.” If approval is unclear, select “No.”
2. Indicate why you (the IRB Analyst) are updating the review. For example: “Analyst setting
ancillary to approved, PRMC approval letter provided by study team”
3. Upload all related approval letters or documentation that indicates the status of the review.
• Select “OK” once all information is confirmed.
Review Types and Requirements:
Device: CHOA
Reviewing organization Device: CHOA
Who ‘approves’ in eIRB? IRB Analyst
Study team actions Contact Anna Alford at CHOA
Analyst actions Follow-up, as needed
Ancillary requirements
Required for studies where all of the following apply:
(1) Children’s Healthcare is a study site
(2) The study includes use of a medical device
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for indication that CHOA is a study location and a device is being studied
Local Study Team Members: If external collaborators affiliated with CHOA
are included: Requirement 1 is satisfied
Study Scope: #2: Does the study evaluate the safety or effectiveness of a
device or use a humanitarian use device (HUD)? If Yes: Requirement 2 is
satisfied
Local Research Locations: If CHOA or a Children’s Healthcare site is listed:
Requirement 1 is satisfied
Device: GA Tech (Legal)
Reviewing organization Device: GA Tech (Legal)
Who ‘approves’ in eIRB? IRB Analyst
Study team actions Provide analyst with confirmation of the completed review
Analyst actions Follow guidance in the pre-review checklist
Ancillary requirements
Required for studies using or evaluating a “home-grown” device from
Georgia Institute of Technology (GA Tech)

Table of Contents
Page 154 of 273
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for indication that a device from GA Tech is used or will be studied
Study Scope: #2: Does the study evaluate the safety or effectiveness of a
device or use a humanitarian use device (HUD)? If Yes: Review is required
Facility: ESJH EJC (Catholic ERD)
Reviewing organization Facility: ESJH EJC (Catholic ERD)
Who ‘approves’ in eIRB? IRB Analyst
Study team actions None
Analyst actions
Send an email to Rebecca Heitkam
requesting an ERD review and attach a
copy of the study protocol and consent form(s) to the email.
Once approval is provided via email, update the ancillary review in eIRB
and upload the approval letter.
Ancillary requirements
Required for all studies where Emory Saint Joseph’s Hospital or Emory
John’s Creek Healthcare are included as study sites.
eIRB locations indicating
the ancillary is required
Local Research Locations: If Emory Saint Joseph’s Hospital or Emory
John’s Creek Healthcare are listed as a site: Review is required
Facility: Grady ROC
Reviewing organization Facility: Grady ROC
Who ‘approves’ in eIRB? N/A: Do not set as “required” (Grady ROC reviews after IRB approval)
Study team actions None
Analyst actions
Notify the study team that they must submit for a Grady ROC review after
the IRB approval is granted.
Ancillary requirements
Review required for all studies where Grady Medical Center or a Grady-
affiliated location is included as a study site, including chart reviews of
Grady medical records.
eIRB locations indicating
the ancillary is required
Local Research Locations: If Grady Healthcare or a Grady-affiliated
location is listed as a site: Review should be triggered
Security: CHOA
Reviewing organization Security: CHOA
Who ‘approves’ in eIRB? IRB Analyst

Table of Contents
Page 155 of 273
Study team actions Contact Carol Price or Jonathan Park
to complete the review
Analyst actions Follow-up, as needed
Ancillary requirements
Required for studies where all of the following apply:
(1) Children’s Healthcare is a study site
(2) The study includes use of a mobile medical application or platform
that collects and stores sensitive, identifiable information
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for indication that CHOA is a study location and a medical application or
platform is used to collect/store sensitive, identifiable information
Local Study Team Members: If external collaborators affiliated with CHOA
are included: Requirement 1 is satisfied
Local Research Locations: If CHOA or a Children’s Healthcare site is listed:
Requirement 1 is satisfied
Security: Emory
*Note: A limited list of applications and software have approval by Emory OIT for research use. If the
study team is only using items included on this list, approval is not required. Approval, even after the Ancillary
Review is requested, will not be issued for this
this list of applications and software.
Reviewing organization Security: Emory
Who ‘approves’ in eIRB? IRB Analyst
Study team actions
Review the OIT Security Review overview and follow the
OIT instructions
for submitting a Security Review.
Study teams should submit a separate security review request for every
platform/application and research study.
Analyst actions
When providing instructions to the study team, add the requested
Security Review to the active list of OIT Security Reviews
.
Once approval is provided, review the review document and verify the
“residual (final) risk” level in each category, as follows:
• If the residual risk is medium or higher: request that the
responsible party for the risk has signed the document
• If all residual risks are low or resolved: no signatures are needed
Ancillary requirements
Required for studies where all of the following apply:
(1) Emory or Grady are study sites (this almost always applies)

Table of Contents
Page 156 of 273
(2) The study includes use of a mobile medical application or
platform not already approved for use and the application or
platform either:
a. collects and stores sensitive and identifiable information, or
b. uses ChatGPT
Note: use this guide
to confirm the ancillary is required
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for indication that a medical application or platform is used to
collect/store sensitive, identifiable information
Security: Grady
Reviewing organization Security: Grady
Who ‘approves’ in eIRB? IRB Analyst
Study team actions
Email the device and data use information to Grady Privacy
Officer D’Andrea Morning
for review and approval
Analyst actions Follow up, as needed
Ancillary requirements
Required for studies where all of the following apply:
(1) Grady is a study site
(2) The study includes use of a mobile medical application or
platform that collects and stores sensitive, identifiable
information
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for indication that Grady is a study location, and a medical application or
platform is used to collect/store sensitive, identifiable information
Local Study Team Members: If external collaborators affiliated with Grady
are included: Requirement 1 is satisfied
Local Research Locations: If Grady Healthcare or a Grady-affiliated
location is listed as a site: Requirement 1 is satisfied
Specialty: Biosafety: EHSO
Reviewing organization Specialty: Biosafety: EHSO
Who ‘approves’ in eIRB? The reviewing organization or the IRB Analyst
Study team actions
Complete a submission in SciShield
(previously BioRAFT) or email EHSO

Table of Contents
Page 157 of 273
Analyst actions Follow up, as needed
Ancillary requirements
Biosafety Program review is required if the human blood, blood products
or tissue is brought to an Emory research laboratory for further
experimentation (not just preparing for shipping elsewhere).
An overview of the review can be found in the
Emory EHSO Institutional
Biosafety Committee (IBC) and Research Health and Safety Committee
(RHSC) Review Process Flow Chart.
eIRB locations indicating
the ancillary is required
Ancillary Review Information: #2: If any items are selected: Review is
required
Specialty: FDA: Controlled Substance
Reviewing organization Specialty: FDA: Controlled Substance
Who ‘approves’ in eIRB? The reviewing organization (Office of Research Integrity and Compliance)
Study team actions
Reference this webpage for the Controlled Substance Checklist
and
information about the approval process
Analyst actions Follow up, as needed
Ancillary requirements
Required for studies that indicate use of a controlled substance in the
protocol or consent.
eIRB locations indicating
the ancillary is required
Ancillary Review Information: #7: Administration of any Schedule I
controlled substances? If Yes: Review is required
Specialty: FDA: REMS
Reviewing organization Specialty: FDA: REMS
Who ‘approves’ in eIRB? The reviewing organization (Office of Research Integrity and Compliance)
Study team actions
Complete the REMS Checklist and contact ORIC
for more information
about the approval process
Analyst actions
Follow up, as needed
Ancillary requirements
Required for studies that indicate a drug will be administered under
the FDA REMS program
eIRB locations indicating
the ancillary is required
Basic Study Information: #1: Review the protocol for discussion of REMS
or a Risk Evaluation and Mitigation Strategy

Table of Contents
Page 158 of 273
Ancillary Review Information: #8: Administration of any Schedule I
controlled substances? If Yes: Review is required
Specialty: HESC (Stem Cell)
Reviewing organization Specialty: HESC (Stem Cell)
Who ‘approves’ in eIRB? IRB Analyst
Study team actions
Complete the Review Application-ESCROC form and email it to ORIC
.
Analyst actions
As soon as the review requirement is identified, contact the IRB Director
or their Designee for further guidance
Ancillary requirements
Required for studies involving Human Embryonic Stem Cells (hESCs) or
Induced Pluripotential Stem Cells (iHPSCs), even if the study itself does
not require (e.g., NHSR because the cells are completely deidentified
before being received by Emory)
eIRB locations indicating
the ancillary is required
Basic Study Information: #1: Review the protocol for discussion of human
embryonic stem cells
Ancillary Review Information:
• #5: Human embryonic stem cells? If Yes: Review is required
• #6: The use of human fetal tissue? If Yes: Review is required
Specialty: Institutional Conflict/IP
Reviewing organization Conflict of Interest
Who ‘approves’ in eIRB? The reviewing organization (Conflict of Interest and Commitment Office)
Study team actions Contact the Conflict of Interest and Commitment Office
Analyst actions Follow up, as needed
Ancillary requirements
Required for studies where there is an institutional conflict of interest or
use of Emory Intellectual Property
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for indication that Emory’s Intellectual Property or an Institutional
Conflict is involved
Waiver Requests and Ancillary Considerations: #3: Is any licensed Emory
intellectual property used in this project? If Yes: Review is required

Table of Contents
Page 159 of 273
Specialty: PRMC (Winship Cancer)
Reviewing organization Specialty: PRMC (Winship Cancer)
Who ‘approves’ in eIRB? IRB Analyst or PRMC Staff
Study team actions Submit a PRMC review application
Analyst actions
Follow up, as needed
Ancillary requirements
Protocol Review and Monitoring Committee review is required for studies
that relate to cancer in any way
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for discussion of oncology or cancer
Ancillary Review Information: #1: Does this study relate to cancer in any
way, even if sociobehavioral, chart review, or secondary analyses only? If
Yes: Review is required
Specialty: Radiation: CHOA
Reviewing organization Specialty: Radiation: CHOA
Who ‘approves’ in eIRB? The reviewing organization (CHOA Radiation Safety)
Study team actions
Complete the Radiation Summary Form
and upload to Local Study
Documents: #3
Analyst actions Follow up, as needed
Ancillary requirements
Required for studies where all of the following apply:
(1) Children’s Healthcare is a study site
(2) The study includes use of radiation or proton therapies at CHOA
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for indication that CHOA is a study location and radiation or proton
therapies are included in the study
Local Study Team Members: If external collaborators affiliated with CHOA
are included: Requirement 1 is satisfied
Local Research Locations: If CHOA or a Children’s Healthcare site is listed:
Requirement 1 is satisfied
Ancillary Review Information:
• #3: Exposure to any radiation? If Yes: Requirement 2 is satisfied
• #4: The administration of any investigational radioactive drugs? If
Yes: Requirement 2 is satisfied

Table of Contents
Page 160 of 273
Specialty: Radiation: EHSO
Reviewing organization Specialty: Radiation: EHSO
Who ‘approves’ in eIRB? The reviewing organization (Emory EHSO)
Study team actions
Complete the Radiation Summary Form
and upload to Local Study
Documents: #3
Contact the Radiation Safety Program with any questions
Analyst actions Follow up, as needed
Ancillary requirements
Required for studies where all of the following apply:
(1) Emory is a study site (this almost always applies)
(2) The study includes any type of radiation, whether scans,
radioactive drugs, or radiation therapy at Emory
eIRB locations indicating
the ancillary is required
Basic Study Information: #3 and #9: Review the protocol and lay summary
for indication that radiation or proton therapies are included in the study
Local Study Team Members: If external collaborators affiliated with CHOA
are included: Requirement 1 is satisfied
Local Research Locations: If an Emory site is listed: Requirement 1 is
satisfied
Ancillary Review Information:
• #3: Exposure to any radiation? If Yes: Requirement 2 is satisfied
• #4: The administration of any investigational radioactive drugs? If
Yes: Requirement 2 is satisfied
Study Team: Conflict of Interest
Reviewing organization Conflict of Interest
Who ‘approves’ in eIRB? The reviewing organization (Conflict of Interest and Commitment Office)
Study team actions
Complete a submission in eDisclose and
contact the Conflict of Interest
and Commitment Office
Analyst actions Follow up, as needed
Ancillary requirements
Required for studies where the PI or a study team member indicates a
Conflict of Interest is present

Table of Contents
Page 161 of 273
eIRB locations indicating
the ancillary is required
Basic Study Information: #7: Does the local principal investigator have a
financial interest related to this research? If Yes: Review is required
Local Study Team Members:
• #1: Review table column titled “Financial Interest.” If Yes: Review
is required
• #2: Review all attachments to see if COI or Financial Interest is
indicated
Waiver Requests and Ancillary Considerations: #3: Is any licensed Emory
intellectual property used in this project? If Yes: Review is required
Study Team: Department
Reviewing organization*
Determine the reviewing organization by searching the
Emory Online
Directory for the Principal Investigator (PI) listed in the eIRB submission.
Check the PI’s Title, School/Division and Department information for their
primary affiliation.
*Note: Instructions and important caveats to this approach are
described below.
Who ‘approves’ in eIRB? The reviewing department
Study team actions Follow up, as needed
Analyst actions Follow up, as needed
Ancillary requirements Required for all new study submissions
*Determining the appropriate Department to review:
• While searching for the PI in the Emory Online Directory, and check the PI’s Title,
School/Division and Department information for their primary affiliation.
o If the PI is not listed in the directory or does not have a department listed, google
their name and Emory University to check for affiliation
o If the directory listing does not align with the research topic, contact Carol Corkran
prior to assigning a departmental review
• If none of the following apply to the details in the Emory Online Directory, assign the listed
“Department” in the directory:
PI Information in Emory Online Directory Organization to assign for Department
Emory + Children's Ped Institute SOM: Peds: Administration
SOMPI: [any] SOM: Peds: Administration
SOM: Peds: Surgery SOM: Peds: Surgery

Table of Contents
Page 162 of 273
SOM: Peds: [anything else] SOM: Peds: Administration
SOM: [non-Peds department] and the
study includes Subpart D
SOM: Peds: Administration
Includes: Pharmacy, Pharmacist, PharmD,
RPh, etc.
EUH: Pharmaceutical Svcs
EHM Pharmaceutical Services EUH: Pharmaceutical Svcs
EHM Specialty Pharmacy EUH: Pharmaceutical Svcs
Includes: RN, Nurse, etc. and
Department is not under SON or SOM
SON: Academic Advancement
SOM: GME: Grad Medicine Educ or
Medical House Staff
Request that PI be replaced with an Emory
faculty mentor as PI and assign to
mentor’s listed department
Emory College (undergrad) or Laney
Graduate School (Masters/Doctoral
students) listings
Request that PI be replaced with an Emory
faculty mentor as PI and assign to
mentor’s listed department
EVP Provost Academic Affairs (and any of
the following offices/units: Central Office,
Academic Innovation, Campus Life, Office
of Diversity, Equity and Inclusion; Office of
Faculty Affairs; Finance, Planning and
Administration; Office of Financial Aid;
Global Strategy and Initiatives;
Institutional Equity and Compliance;
Michael C. Carlos Museum; Research
(Office of SVPR); Office of Undergraduate
Admission; Office of Undergraduate
Affairs; and Office of University Registrar)
EVP Academic Affairs & Provost
All other non-academic departments
Contact an IRB Assistant Director to clarify
the appropriate reviewing organization
Notes for assigning the reviewing organization:
• The directory generally lists three levels of information for the Department, each separated
by a colon (”:”). Unless otherwise specified, the ancillary should be determined based on the
first two statements and “Administration” as the final group.
• Example: A study PI in the “SOM: HMO: Hematology” department will need their study
reviewed by “SOM: HMO: Administration” in eIRB.
• If the study PI is also the only Departmental Approver, re-assign the ancillary review to the
department’s “Parent Organization” as follows:
Conflicted PI’s Department Organization to assign as Department
ECAS: [department name] ECAS: Dean of the College

Table of Contents
Page 163 of 273
SPH: [department name] SPH: Dean’s Office
SOM: Medicine: [department name] SOM: Medicine: Admin
SOM: [department name] SOM: Dean’s Office
SON: Academic Advancement SON: Dean’s Office
Study team: S-I Advisory
Reviewing organization Study team: S-I Advisory
Who ‘approves’ in eIRB? The reviewing organization (Office of Research Integrity and Compliance)
Study team actions Reference this webpage for more information
Analyst actions Follow up, as needed
Ancillary requirements
Required for studies where Emory faculty, staff, etc. are listed as the IND
or IDE holder for a study drug or device
eIRB locations indicating
the ancillary is required
Basic Study Information: #1: Review the protocol for discussion of a
sponsor-investigator.
Drugs and Devices: Review all IND or IDE documents and see if any
persons listed on the applications are on the local study team.
Conflict of Interest ancillary review is required, scrutinize the application
to confirm if an S-I review is also needed.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2023-02-24
Added reminder about security review
2023-07-06
CHOA clarity and archived changes over a year old
2023-09-11
Removed Office of Quality
2023-09-19
Clarity on CHOA Device ancillary review
2024-04-11
Substantial updates including: Added instructions for Pre-Review selections; Added detailed instructions for
each ancillary review; Added ancillary reviews for security reviews, device legal review, and delineated
requirements for reviews based on the involved institutions (CHOA, Emory, Grady)
2024-08-15
Added other offices/units that EVP Provost Academic Affairs provides departmental approval review.

Table of Contents
Page 164 of 273
SOP Title:
eIRB Processing of New Study Applications- Preliminary Analysis through Approval
SOP
Study Management
Established:
03/12/2015
Last Revision:
9/11/2023
IRB Staff Initial Analysis/Screening
Note: Find all the checklists on this SOP in this folder: H:\General\Admin IRB Documents\Checklists-
Staff, Forms, and Templates\1. Staff Screening Checklists and Tip Sheets.
1. Once assigned as the IRB coordinator, it will appear in the “Pre-Review” state under your inbox. For
more information about pre-review and ancillary review information, review our SOP
.
2. Open the appropriate checklist (non-exempt, exempt, retrospective)
3. While in the main study workspace, click the “View Study” button. Screen the study per whichever
checklist you chose.
a. This form is not purely a checklist; you are expected to write notes to yourself for posterity and
notes that can be easily transformed into suggested revisions for the study team.
b. Manage applicable ancillary reviews including department review.
4. Once done screening, click the “Submit Pre-Review” button in the main study workspace. Look at
the Huron Staff Quick Reference
(page 17) for additional information on how to complete this
process.
a. For any study that requires HIPAA authorization and/or waivers, you MUST check the box for
OCR (Office of Civil Rights) in question 1.
b. Check applicable boxes in question 2 and fill out any applicable
determination/waiver/population worksheets.
c. Check one or more boxes in question 3 to best characterize the study.
d. Check any boxes that apply in question 4.
e. Enter the missing materials in question 5.
f. Enter any other notes, including requested changes to the submission, in question 6.
g. Upload the new study checklist in question 6.
h. Check “No” in question 7 unless the study is ready to be reviewed as submitted, with no
revisions.
i. Click OK.
If study need revisions after your initial review
1. Click “Request Pre-Review Clarifications” button to send back to study team for revisions. Look at
the Huron Staff Quick Reference
(page 15) for additional information on how to complete this
process.
a. You are required to remind study teams of their changes to the submission, at least at a weekly
basis. If they are not responding to your comments or emails, contact a Director to withdraw
the study

Table of Contents
Page 165 of 273
b. Follow up with the study team if a response is not received in a timely fashion. If a submission is
in requested changes for more than 30 days with no answer from the study team, the analyst
should withdraw it. The analyst needs to document their efforts in contacting the study team
and email them before withdrawing the study.
2. Once re-submitted, screen the submission for the changes you requested. If the study team made
the revisions to your satisfaction, click “Submit Pre-Review” again.
3. Make any necessary changes to the responses in questions 1-7 and change the response to question
8 to “Yes.” This will change the state to “Pre-Review Completed.”
Ancillary Reviewers
1. Click the “Manage Ancillary Reviews” button and click the “Add” button to add any appropriate
reviewing bodies. Select the group (not person) under question 1, select the group again under
question 2 and click on yes if a response is required.
2. Review this page
(under Submit a study in eIRB) on our website or the checklist for more
information about ancillary reviews.
Finish Pre-Review
1. If going the full board route, complete your Pre-Review and include notes that would be helpful to
have during the meeting, such as why the study might require FB review (if not readily apparent),
special determinations, subparts, potential IRB member conflicts, etc. Fill out the last page of the
non-exempt checklist for this purpose.
2. Assign the study to a Designated Reviewer or full board meeting agenda.
3. Note that the pre-review can be edited after it is initially submitted in all states before a letter is
prepared in post-review.
Designated Reviewer route:
1. Before sending to a reviewer ensure that:
a. All ancillary reviews are completed, including department review.
b. All CITI/Intro to Research training is complete for all study team members
c. If external staff is being added, that the IAA is completed (if not, the staff member can be
removed and later added via a modification)
2. Select a reviewer in question 1 according to our current SOP
on who can review what.
3. In question 2, enter notes to the reviewer such as—but not limited to—exempt/expedited
categories, subparts, recruitment methods, verbal/online vs. signed consent, HIPAA waivers, funding
source, and whether the study has the appropriate permissions/ethics approvals in place.
4. In question 3, upload any pertinent checklists:
a. Emory Subpart B Checklist
b. Emory Subpart C Checklist
c. Emory Subpart D Checklist
d. Emory Checklist- Cognitively Impaired Subjects

Table of Contents
Page 166 of 273
e. Make sure other appropriate checklists (HIPAA, IND exemption, Device Checklist, REMS,
Controlled substance, Dietary supplements, Mobile medical Apps, etc.) are in the record as
filled by the study team. These checklists are located on our website under “
Study
Submission Guidance)
5. Click OK. This will change the state to “Non-Committee Review.”
6. Once the reviewer submits their review, the state will change to “Post-Review.”
7. If DR requests revisions:
a. First, consider whether the revisions would modify any of the findings of the pre-review (federal
agency involvement, waivers, etc.) Make the pre-review edits necessary to reflect the changed
status of the study by clicking “Edit Pre-Review.”
b. Click “Prepare Letter” in the main study workspace.
c. Choose the appropriate template and click “Generate.”
d. Under “Draft letter,” click the link to download the letter template and make revisions.
e. You will need to save a local copy of the letter, make the revisions within that letter, and upload
the revised copy to the study using the “Upload Revisions” option in the activity details.
f. Once revisions are complete, click “Send Letter.”
g. Follow up with the study team if a response is not received in a timely fashion. If a submission is
in requested changes for more than 30 days with no answer from the study team, the analyst
should withdraw it. The analyst needs to document their efforts in contacting the study team
and email them before withdrawing the study.
h. After the PI/proxy submits the changes, the study will appear in the “Modifications Submitted”
state.
i. Click the “Compare” button in the left-hand top corner of the main study workspace to examine
the changes to the submission made by the study team. Look at the
Huron Staff Quick
Reference (page 16) for additional information on how to complete this process.

Table of Contents
Page 167 of 273
j. In the Basic Study Information section, question 8, click the “History” link under “Document
History.”
k. On that pop-up page, check the boxes for the documents you want to compare, and then click
the “Compare” button; this will produce a “track changes” copy of the new protocol. Screen the
document for the revisions you requested as well as any other revisions the study team may
have made. Once done, click “Exit.”
l. If the changes requested by the reviewer were not adequately addressed, click the “Review
Required Modifications” button.
i. A new window will open (see left)
ii. Confirm whether the study requires continuing review and make sure that it is reflected in
question 1 on the pop-up page.
iii. Enter the approval date (and expiration date if applicable) in question 3.
iv. Enter any relevant notes in question 4 and upload any documentation in question 5.
v. Click no on Question 6. This returns the study to the team for further action. Repeat this
process until all changes are adequately addressed
m. If the changes look complete, click on “Assign to Designated Reviewer” so they can review the
contingency review.
n. After the designated reviewer has submitted their review, ensure that the approval/expiration
dates, especially after a pending review, are correct. You can submit a review yourself to
correct the issue, adding a comment of why you are resubmitting. If unsure, ask one of the TLs.
i. If you are ready to finalize approval, first review the expiration date in the designated review
space. Ensure that the expiration date is the overall study expiration date.
1. If the expiration date is incorrect, click on “submit designated review” and correct it.
8. Check once more that all Ancillary Reviews are complete.If not, final approval must be held until
complete.
9. After this check, click “Finalize Documents” on the study worksplace. Select the documents that
need to appear in the approval letter and click OK. The Documents tab on the study workspace will
include links to the final versions of the documents.
Full Committee Review route:
1. Look at the Huron Staff Quick Reference
(page 23) for additional information on how to complete
this process. Click the “Assign to Meeting” activity and assign the study to the next available,
appropriate meeting; this will change the state to “Committee Review.”
2. Click on “Edit Preview” to include an updated NS checklist and update the board before the meeting.
This should happen the Friday before the meeting.
3. Keep in mind the membership of the committee.
4. Try not to assign studies where PI/Co-Is are members of that committee but try to assign studies to
committees where the specialty of the study is represented in the membership.
5. Reviewers will be assigned to the study by the meeting pod.
6. After the study is reviewed:
a. Study was approved as is: prepare letter as detailed in the next section.

Table of Contents
Page 168 of 273
b. Pending or Deferred:
i. Click on prepare letter and select the appropriate pending letter. All pending/deferred items
should populate in the letter. Reconcile the letter with the notes in the “reviews” tab to
confirm all the information is in the letter. Send the letter to the team.
ii. When the study team submit the changes
1. If pending:
a. Confirm that all ancillary reviews are complete (except Office of Quality, which is not
required to be completed in our system).
i. If any Ancillary Committees have required changes to the
protocol, those changes should be reviewed at the next
available full board meeting instead of being reviewed by
the Chair. Notify your Team Lead and note rationale in Pre-
Review.
ii. If the Ancillary Committees require revisions to the consent
form based on their established template language, those
changes do not need to return to full board.
iii. COI management plans must be fully approved by IRB’s
CoRe, and required changes made to consent form and
smartform, before final approval by Chair. These changes do
not require full board review.
b. Review the study team’s changes.
i. If Cost Option was completely unknown at initial review,
and comes back as Option 3 (i.e. participants will have to
pay for research-driven items or procedures), alert your
Team Lead as the IRB may not approve.
ii. If other changes were made beyond what was required by
the full board (e.g. a new protocol version from Sponsor),
assess whether the changes would require expedited or full
board review, if they had come in as a stand-alone
modification.
c. to ensure completeness, email the Chair of the FB meeting with a
link to the study. Ask the Chair to not click on “Review Required
Modifications”. Instead, they can email you back letting you know
the changes were adequate or they can log a private comment. If
the Committee requested, the link should also be sent to another
reviewer along with the Chair.
d. If the Chair is completely unavailable for more than five business
days: Contingency reviews may be referred to a Co-Chair who was
in attendance. If none available, it may be referred to the primary or
secondary reviewer of that item as long as they were in attendance.
(e.g. Dr. Stein), and if none, it may be referred to primary reviewer.

Table of Contents
Page 169 of 273
e. Click on “Review Required Modifications”. The “approval date” would be the date of
the meeting. The “effective date” is the
date the vicechair confirmed the
changes were adequate and the study
can receive final approval.
f. After the above is completed,
send the approval letter (follow the next
section).
g. Follow up with the study team
if a response is not received in a timely
fashion. If a submission is in requested
changes for more than 60 days with no
answer from the study team, the analyst
should withdraw it. The analyst needs to
document their efforts in contacting the
study team and email them before
withdrawing the study.
2. If deferred:
a. After reviewing the changes to ensure completeness, click on “submit pre-review”.
Document any changes to your review, if pertinent. Assign to the same committee
who review the study.
b. Follow up with the study team if a response is not received in a timely fashion. If a
submission is in requested changes for more than 60 days with no answer from the
study team, the analyst should withdraw it. The analyst needs to document their
efforts in contacting the study team and email them before withdrawing the study.
Preparing approval letter
1. Look at the Huron Staff Quick Reference
(page 25) for additional information on how to complete
this process. After all the changes has been confirmed go to the main study workspace, click
“Finalize Documents.” Make sure you do not skip this part, as this is step will allow the approved
documents to populate in the letter.
2. On the pop-up page, select the documents you wish to finalize, and click “OK.”
3. Click “Prepare Letter.”
4. Under “Draft letter,” click the link to download the letter template and make revisions.
5. You will need to save a local copy of the letter, make the revisions within that letter, and upload the
revised copy to the study using the “Upload Revisions” option in the activity details.
6. Once revisions are complete, click

Table of Contents
Page 170 of 273
7. Click “Send Letter.”
8. Study will now appear in “Approved” state.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/4/2021
Fix errors in designated review and changes in the contingency review process.
04/06/2022
Julie made some updates – see SOP archive for changes
4/22/2022
Updated location of checklist and archived updates over 1 year old
9/11/2023
Contingency review clarifications

Table of Contents
Page 171 of 273
SOP Title: Naming Conventions for eIRB Studies
SOP Category:
Study Management
Established:
10/30/2017
Last Revision:
10/23/2020
PURPOSE
The purpose of this document is to describe the naming conventions used to signify special study
designations in the short title of the study.
DEFINTIIONS / NOMENCLATURES
• Emory Sponsor Investigator study – [SI]
• External IRB shell – [XIRB]
• NCI Central IRB – [CIRB]
• Department of Defense – [DoD]
• Veteran Affairs – [VA]
• Western IRB – [WIRB]
• REMS- [REMS]
• Emory as Single IRB [sIRB]
PROCEDURE
When reviewing a study that meets one or more of the special study considerations listed above, edit
the study short title to add in the appropriate bracketed designation before the official study short title.
If more than one designation applies, list multiple designations in alphabetic order.
When adding the nomenclature to the study title, log a comment to the study team explaining that the
IRB added that information for future study tracking.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/21/2018
Added REMS as a nomenclature to use
6/21/2019
Removing non-used options
2/11/2020
Added/removed options
10/23/2020
Added/removed options

Table of Contents
Page 172 of 273
SOP Title: RDRC Studies
SOP Category:
Study Management
Established:
12/09/2015
Last Revision:
12/09/2015
PURPOSE
The purpose of this document is to describe the procedures for the coordination between the
Institutional Review Board (IRB) and the Emory Radioactive Drug Research Committee (RDRC) on
protocols involving the use of radioactive drugs for research projects designed to obtain basic
information regarding metabolism (e.g., kinetics, distribution, and localization) or human physiology,
pathophysiology, or biochemistry.
BACKGROUND
The Radioactive Drug Research Committee (RDRC) program under 21 CFR 361.1
permits certain basic
research using radioactive drugs in humans without an IND. The RDRC is the body charged with
classifying all radioactive drugs as either new drugs requiring an Investigational New Drug Application
(IND) for investigational use (21 CFR 312), or as generally recognized as safe and effective when
administered under the conditions specified in the RDRC regulations. Key requirements include that 1)
number of subjects should not exceed 30, 2) only adults with legal capacity be enrolled, 3) all females of
childbearing potential either confirm they are not pregnant on the basis of a pregnancy test or state in
writing they are not pregnant, and 4) the investigator shall immediately report to the RDRC all adverse
effects associated with the use of the radioactive drug.
RESPONSIBILITIES
• RDRC- Review and approve the use of research-related administration of radioactive material to
subjects. RDRC is also tasked with reviewing all adverse effects associated with the use of the
radioactive drug in research and immediately reporting to the FDA all adverse reactions probably
attributed to the use of the radioactive drug in research.
• IRB – Ensure human research protocols involving the research-related administration of radioactive
material to subjects have prior RDRC approval and that the study protocol include the required
reporting to RDRC. In addition, alert RDRC and study team of need for IND if study team requests
increase in enrollment to over 30 subjects obtaining the radioactive agent.
PROCEDURE
1. When a study is submitted that involves a radioactive tracer not approved by the FDA for the
indication described in the study, the IRB analyst will review whether the study has an IND or RDRC
approval for the use of the tracer.
a. If the study does have an IND, than the study falls outside the scope of this SOP.
2. For studies that meet the qualification for RDRC IND exemption, the IRB analyst will ensure that:
a. RDRC approval has been granted before the IRB grants final approval.
b. The authorized investigators in the RDRC approval letter are listed as study staff, including
the “Authorized User”
c. The radioactive tracer is listed in the drug section of the eIRB Smartform

Table of Contents
Page 173 of 273
d. The protocol and DSMP include that the investigator shall immediately report to the RDRC
all “adverse effects” associated with the use of the radioactive drug in the research study.
e. The protocol and consent note that only adults (18 and older) with legal capacity will be
enrolled.
f. The protocol and consent note that all females of childbearing potential confirm they are
not pregnant.
3. Once all of the above requirements have been verified, the IRB analyst should assign the study to an
IRB Committee meeting – these studies do not qualify for expedited review under F(1) due to the
possibility of allergic reaction.
4. After the initial IRB approval, IRB analyst should note whether future modifications that
substantially change the protocol may require additional review by the RDRC or, alternatively, an
IND application. Analyst should consult with RDRC if needed.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 174 of 273
SOP Title:
Translation of Informed Consent Documents
SOP Category:
Study Management
Established:
11/17/2016
Last Revision:
08/15/2024
PURPOSE
To instruct analysts about acceptable methods of translation of written informed consent documents
into languages other than English.
SCOPE
Applies to all non-exempt studies that plan to target non-English speakers for enrollment, regardless of
whether the study involves international sites. This does not apply to studies requesting to use a “short
form” to enroll occasional non-English speakers. This SOP also does not apply to exempt research (see
exempt screening checklist for relevant requirements). The Emory IRB will only review the research
activities that Emory agents are engaged in. If consent documents fall outside of the scope of Emory IRB
review, there is no need for us to review translations. However, if Emory is the primary awardee of a
grant, translated documents will likely be required for IRB review, regardless of whether Emory is
otherwise involved in the human subjects’ research activities.
INTRODUCTION
In order to ensure the quality of translation of consent documents into foreign languages, the Emory IRB
requires some level of documentation that the translation is accurate.
Recommendations for study teams:
• In most cases, it’s best for the team to first seek approval of an English version of the informed
consent document before beginning translation to prevent having to revise both the English and
translated documents based on IRB change requests. The translations would then be submitted
via Modification. The English version of the informed consent document should be stamped
after initial approval.
• A local Ethics Committee or IRB may require changes to translated informed consent documents
that are submitted for their approval. For this reason, it may be advantageous for study teams
to wait to submit their translated documents to the Emory IRB until after the local approval is in
place.
• Note: Emory may collaborate on studies led by an international site where the informed
consent document was first created in a foreign language. In that case, the translation
policy still applies, but the translation would be into English.
There are two acceptable methods for documenting the quality of the translation
Methods of Documenting Translation Quality:
• Certified Translator: The team can hire a certified translator to translate the English version of the
form into the foreign language. As proof of translation by a certified translator, the study team
may submit:

Table of Contents
Page 175 of 273
o Current, valid certification of translator’s credentials along with an invoice or memo
stating the specific document that was translated.
o Other: Consult with your Associate or Assistant Director or the Director any other
form of documentation that mentions the specific document that was translated
and attests to the translation quality
In addition to the documentation for translation services, the IRB must receive the translated
forms.
• Translation by a non-certified translator: This is a two-step process. To start, anyone, including
members of the study team, may translate the English version of the informed consent document
into the foreign language. For the back-translation, the study team must find someone else to
perform a translation into English from the already-translated foreign language document. The IRB
reviewer will compare both English documents to confirm that the back-translation retains the
same message as the original English version. All versions, appropriately labeled, should be
submitted to the IRB.
PROCEDURE
1. An analyst receives a new study or an Modification that indicates intent to enroll
participants that do not speak English (should be evidenced by selecting “Non-English
Speakers” as a Study Population type), along with translated versions of consent documents.
2. The analyst reviews the new study or Modification to see if documentation of one of the
appropriate translation methods has already been provided.
3. If documentation hasn’t been provided, the analyst should log a comment to notify the
study team that they should begin the process of documenting translation through one of
the above-listed methods and that translated copies will not be approved until that is
complete, though the rest of the study may be approved. The analyst should continue triage
of the new study or Modification.
4. The analyst assigns new study or Modification to a designated reviewer or to a full board
agenda. The analyst makes a note in the study history as to the current status of translated
documents.
a. If adequate documentation has not been provided: The study or Modification can
be approved without documentation of translation, but the study team will not be
able to enroll any subjects using the translated consent form until they provide
documentation of one of the above-listed methods of translation.
The IRB analyst should not stamp the foreign language consent forms until a
reviewer is able to review the documentation of translation.
The IRB analyst should notify the team in the approval letter that the foreign
language consent forms are not approved for use, and furthermore, that the team
cannot enroll any subjects using translated consent documents. Also, the analyst
should notify the team that an Modification will be required to submit
documentation of translation.
5. Once the new study or Modification has been approved, the analyst lists all of the consent
documents in the approval letter. The analyst should stamp all approved versions of the
informed consent documents, including the English version even if it’s not going to be
presented to subjects.

Table of Contents
Page 176 of 273
Note: Sometimes the formatting and language of translated versions of documents look
strange after eIRB applies the stamp. Most often, teams are able to open the documents and
see the correct formatting and language with the stamped approval date.
Modifications revising approved, translated ICFs
1. Modifications that are only making administrative changes (such as contact information) can
be approved without further translation quality documentation as long as the change can be
verified in the revised document.
2. Minor Modifications that can be sent to a staff designated reviewer can receive pending
approval with only a tracked changes version of the English consent form. The Modification
would be ready for contingency review with a translation of the revised English form and
documentation of the translation quality through one of the two approved methods.
3. Modifications that may change the risk/benefit ratio of the study and would require
designated review by a vice chair or full board review may require two Modifications to
update the study and the translated ICF. One Modification would include the protocol or
other study changes and incorporation of those changes into the English version of the
consent document. After approval of those changes, the study team would follow up with
translation of the revised English ICF and documentation of translation quality through one
of the approved methods.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/1/2019
It was clarified that the English version of the consent form should be stamped when approved.
08/15/2024
Clarified scope

Table of Contents
Page 177 of 273
SOP Title:
Training Verification
SOP Category:
Study Management
Established:
10/18/2010
Last Revision:
8/15/2024
PURPOSE
The purpose of this SOP is to guide IRB staff how to verify that study team members have current CITI
certification(s) required for the type of study.
SCOPE
This SOP applies to all new studies, all Continuing Reviews and Modifications that are adding study
personnel.
DEFINITIONS
• CITI – the Collaborative Institutional Training Initiative
RESPONSIBILITIES
• IRB analyst: responsible for checking that the research team staff has received the required training
for new submissions, Continuing Reviews, and Modifications (adding new staff)
• Office for Clinical Research: responsible for answering study team and IRB questions about clinical
trial training requirements other than those pertaining to biomedical and sociobehavioral CITI
training
NOTE: Study team members who are external to Emory must complete their home institution’s CITI
training requirements unless they are not affiliate with another institution.
PROCEDURE
1. Determine the CITI training required for the study. Review the guidance on the Emory IRB website
under Resources, Training, Courses
as well as the lay summary, scope and clinical trial questions in
the IRB submission. For continuing reviews and modification, review the most recent pre-review.

Table of Contents
Page 178 of 273
2. A new window will open. (See example below with names blocked out.)
3. Verify that all required CITI modules appear for the study team members you are verifying. For new
studies and continuing reviews, confirm CITI is current for all study team members. For
modifications, only confirm CITI is current for those study team members that are being added).
• If current CITI training is missing for a study team member, instruct the study team to have that
individual complete the required training. If the person’s training is current, but does not populate
in the View CITI Training tab, ask the study team to log a comment with the CITI training certificate
(not the transcript).
• For continuing reviews, if a study team member (other than PI or a Co-I who has expertise required
for the study) does not have current CITI training, and the study is close to the expiration date, let
the study team know they can opt to remove the study team member temporarily and add them
back via a modification once the continuing review is approved. The team will need to submit a
modification to remove the study team member and this modification needs to be approved before
approving the continuing review. The modification can be reviewed by any IRB staff by following the
Adding/Removing Study Personnel SOP.
• You can assign a submission to a full board meeting with CITI training pending, but you cannot
assign to an expedited reviewer until all study team members have current CITI training.
Procedure for Member Search in CITI (for those who have Admin access only)
• Go to www.citiprogram.org
and click “log in through my organization” or log in using your own
personal login.
• Click “Admin” button at the top of screen.
• Click “Members” and then “Member Search.”
• Enter last name (and first if desired) where indicated to search for the member.
• Click on the member’s name.

Table of Contents
Page 179 of 273
• Click “Course Completion History” on the right.
• Click the link under the column “Completion Record” to view or save the certificate.
REFERENCES
• Emory Required Training for Investigators, Clinical Research Nurses, and Coordinators Chart
.
• CITI Program modules: www.citiprogram.org
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
12/11/2020
Added that CHOA staff members should comply with OCR training requirements
3/11/2021
Added information from website about CITI expiration
8/30/2021
Updated CHOA and archived log of updates older than 1 year
7/05/2022
Updates for clarity
8/15/2024
Updates to remove key concepts, external teams complete their own institution’s CITI training

Table of Contents
Page 180 of 273
SOP Title:
Electronic documentation of informed consent via “electronic signature” or “digital
signature”)
SOP Category:
Study Management
Established:
12/20/2016
Last Revision:
3/11/2021
PURPOSE
The purpose of this document is to explain the use and IRB requirements for the currently available
options Emory researchers have to use electronic informed consent documentation (eICD) after
consenting a subject into a study using (or not) an electronic informed consent method.
This SOP is not applicable for cases where the IRB can waive written signature/documentation of
consent (e.g. online survey studies) or for studies done at the VA or CHOA, as they do not need to obtain
signature after online consent
Note: HIPAA authorization may be obtained via electronic signature as well, when in compliance with
the below SOP and federal guidance. OIT will not review requests of eICF if the study does not involve
IIHI, PHI, or sensitive information but the IRB will need to verify that the app or software. For FDA
studies, the IRB needs to ensure that capturing the subjects’ signature after consent complies with the
requirements in Part 11.
SCOPE
The SOP applies to all studies submitted to the Emory IRB and when the Emory IRB provides local
context information to external IRBs.
RESPONSIBILITIES
• IRB analyst – responsible for letting the study team know about the currently available options for
the use of eICD, making sure the use aligns with previously approved parameters given to us by LITS
• OIT representative - reviews proposals to implement eICD outside the currently approved options
• Team Q- facilitates the discussion with OIT representatives and investigators
FEDERAL GUIDANCE (OHRP and FDA)
• http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guid
ances/ucm436811.pdf
PROCEDURE
• Review our guidance document (When is an OIT security review needed?) to verify if the current
software needs LITS review.
o Electronic documentation of consent is not permitted the AVAMC (though they do allow
online consent with a waiver of the signature when regulatory criteria are met). Please
verify with the IRB Directors if this is still the case when reviewing your study as
exceptions may have been allowed due to public health needs.

Table of Contents
Page 181 of 273
o Electronic documentation of consent is allowed at CHOA only if using REDCap. For other
platforms, the study team will need to put a request for security review by the CHOA IT
group.
• The protocol and/or smartform should include a plan for providing copies of the signed consent to
participants. (HHS and FDA regulations require that the person signing the informed consent be
given a copy of the written informed consent form (45 CFR 46.117(a) and 21 CFR 50.27(a)) unless
the requirement for documentation of informed consent has been waived under 45 CFR 46.117(c)
and 21 CFR 56.109(c)).
o Although FDA regulations do not require that the subject’s copy include a signature, FDA
recommends that a copy of the signed informed consent form that includes the date when the
eIC was signed be provided to the subject.)
o The copy provided to the subject can be paper or electronic (i.e. be provided on an electronic
storage device, not via email unless encrypted). If the copy provided includes one or more
hyperlinks to information on the Internet, the hyperlinks should be maintained and information
should be accessible until study completion (if a paper version is provided, it should contain the
necessary content from any hyperlinks).
• The protocol and/or smartform must include a plan for verifying the identity of the subjects that will
be electronically signing the Informed Consent, for FDA-regulated investigations.
o FDA regulations do not specify any particular method for verifying the identity of an individual
and accept many different methods. For example, verifying someone’s identity can be done by
using information from some form of official identification, such as a birth certificate,
government-issued passport, or a driver’s license. Also, the use of security questions to confirm
an individual’s identity can be considered.
Redcap
• Besides adding the intention of using Redcap in the study protocol, the study team should submit to
the IRB copies of all forms (electronic and paper forms) and informational materials, including any
videos and Web-based presentations, which the subject will receive and view during the eIC
process.
• The study team should submit an MS Word version of the informed consent language that will be
signed electronically by subjects
• Once IRB-approved, the study team should not send the consent form to subjects via email, unless
encrypted. If not using the encrypted email they should explain in the submission that the form will
be sent via a link to an email previously provided by the study subject or LAR.
• The signature area could be drafted the same as the ICF/HIPAA template or could allow for
documentation of the signature of the person obtaining consent at a later time. Here is an example:
___________________________________ _________________________________
Name of Person Conducting IC discussion Date when IC discussion took place
___________________________________ __________________________________
Signature of Person Conducting IC discussion
Date when IC was signed by person obtaining consent
• The Redcap system must capture the signature of the subject in a way that it can be electronically
audited. The ideal format is as follows:
Table of Contents
Page 182 of 273
1)
Name of Patient
* must provide value
2)
Name of Legally Authorized
Representative with authority for
research decisions
* must provide value
3)
Relationship to Patient
* must provide value
4)
* must provide value
Add signature
5)
Current Date and Time
* must provide value
NowM-D-Y H:M:S
Please press the NOW button
• The study team should later submit, via Modification, a final electronic form to the IRB to show that
the eICF version contains the same information as the mock-up approved by the Emory IRB,
including document approval and version date. This can be accomplished by sending a link to the
consent form for our review. The study team cannot enroll anyone with the electronic method until
this Modification is approved.
• The study analyst should include in the approval letter the following:
o The IRB approved the use of electronic informed consent via Redcap is contingent upon the
submission of a Modification showing the final version of the electronic informed consent as it
will be seen by subjects
o Do not use the eICF until this Modification is approved by the IRB
o Per Emory’s sensitive information policy/HIPAA (as applicable), do not send the copies of the
informed consent to subjects via email. You may send a link to the system containing the eICF
for the subject’s review. The Modification can be sent for expedited review.
See below an example of an acceptable electronic ICF in Redcap:

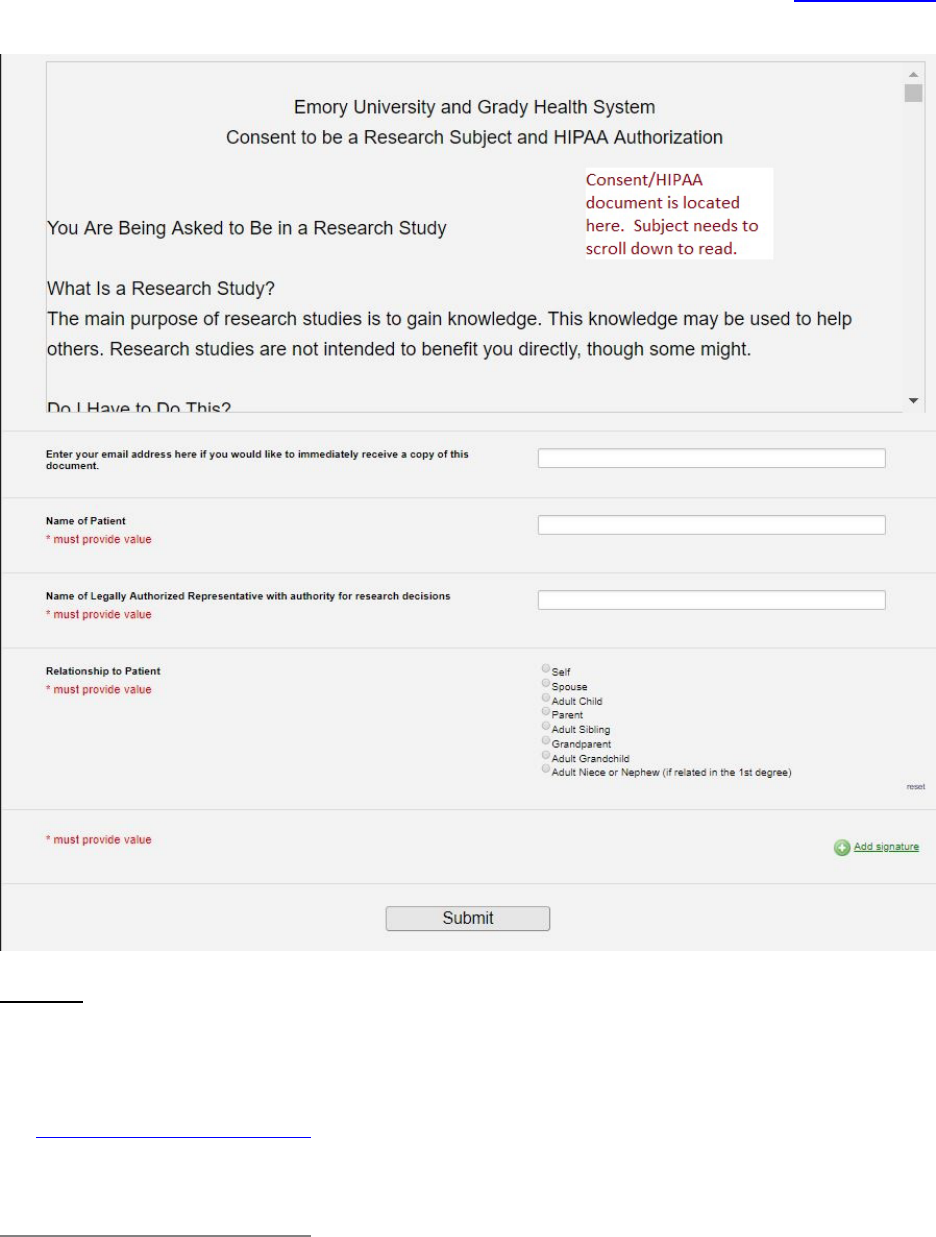
Table of Contents
Page 184 of 273
DocuSign
• Because DocuSign only adds signature lines to current documents, the study team can obtain
approval of their documents with initial approval of the consent. The study team should add to
their protocol there are using DocuSign for eIC. If the study is using DocuSign for an FDA regulated
study, the team should say in the protocol that the DocuSign they are using is part 11 compliant.
Send this link with information
about the process to obtain the account to the team when asking
about this. This information includes instructions of how to transmit to the FDA directly but the
relevant part about obtaining a Part 11 compliant account if found here:
Getting Your Emory DocuSign Account
• In order to submit e-documents to the FDA’s electronic submissions gateway (ESG), your Emory NetID will
need to be added to the FDA-compliant Emory DocuSign account.

Table of Contents
Page 185 of 273
• Go to emory.service-now.com
• Click on the “Request Something” link on the Service Now homepage
• Use the search field to type “FDA Document Transmission”
• Fill out the available form to request your NetID be added to the FDA-compliant DocuSign account
• You will be contacted by a member of the LITS WebGroup once your request has been processed
• Note: if something unexpected occurs that prevents you from accessing the Service Now request form, you
can send an email to webgroup@emory.edu
to have your request processed. Be sure to include your NetID,
your Emory department/school, and the business reason for your request in your email.
Logging in to DocuSign
• Go to docusign.emory.edu
• Log in through the Emory Login page
• If you are working remotely, you may be required to authenticate using Duo.
• If you do not yet have Duo installed, follow the setup prompts to do so.
If you have an account in both the general purpose and FDA CFR Part 11 account, you will need to switch
accounts. To do so, click on your initials or profile in the upper right-hand corner. Select "Switch Account" or
verify you are in the FDA account. Under your name and email, you should see an account name of "Emory
CFR 21 Part 11 Compliant".
Unapproved OIT software
• If the study team is not using an approved eConsent method, they should submit a ticket for an OIT
security review of the app/software they want to use for eConsent.
• After asking the team to submit an OIT ticket, add the information to this spreadsheet
.
• The study team should go to this link: https://emory.service-
now.com/sp?id=kb_article&sys_id=e5c2cd88f5cdf1c055c77bd8604896c2 to place a ticket for this
review.
• The form has the information to provide under “more information”
• The study team should be advised that this process may take time, and to work with OIT and let the
IRB know if their eICF platform was approved.
o If the study is funded, let OSP know (at osp@emory.edu
) that the study will be reviewed
by OIT for a security review as this may affect contract negotiations or require additional
actions such as a Business Associate Agreement.
• For more information about the OIT security review report, and how to address its findings, see
guidance found in this folder
.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
1/20/2017
Updated process when not using Redcap
4/20/2017
Updated broken links
5/17/2018
Remove the need for the study team to send email to Derek or Maria
7/25/2018
Added that we should let OSP know if the study is under a security review when the study is funded
4/11/2019
Adding example of eICF and clarifying when this request needs to be sent to LITS
4/14/2020
Clarified that email can be used if it is encrypted. Other minor clarifications.
5/18/2020
Clarified that, for FDA regulated studies, a part 11 compliant platform is required for eConsent.
Clarified other parts of the SOP and added a link to an excel sheet we need to update each time a
new LITS request is asked from the study team.


Table of Contents
Page 187 of 273
SOP Title: Mobile Devices and Mobile Medical Apps Used in Research
SOP Category:
Study Management
Established:
12/20/2016
Last Revision:
5/24/2020
PURPOSE
The purpose of this SOP is to inform analysts, in a step-by-step fashion, how to identify and process
studies that are using mobile devices and apps, including the use of mobile medical devices and apps.
SCOPE
The SOP is intended to cover two types of research studies:
• Studies that use mobile devices for communication/data collection with subjects (e.g. smartphones,
tablets) regardless of what specific applications are used on those devices. The main issue is data
security/privacy.
• Studies that are testing or using applications (‘apps’) on mobile platforms that meet the definition of
“medical device” per FDA. The main issue is whether FDA regulations apply, and if so, which ones.
Data security is also a possible issue.
DEFINITIONS
• FDA Enforcement Discretion: The FDA does not intend to enforce the requirement of the FD&C Act.
• Medical Device: An instrument being used for the diagnosis of a disease or other conditions, or the
cure, mitigation, treatment, or prevention of disease, or is intended to affect the structure or any
function of the body of man.
• Mobile App (Application): A software application that can (but not inherently is) run on a mobile
platform. It can also include web-based software applications executed on a server.
• Mobile Medical App: Any mobile app that meets the definition of a device, in section 201(h) of the
Federal Food, Drug, and Cosmetic Act; and is intended to:
o Be used as an accessory to a regulated medical device; or
o To transform a mobile platform into a regulated medical device.
• Mobile Platform: Commercial, “off-the-shelf”, computing platforms which are handheld in nature
[e.g. smartphones, tablets, portable computer devices].
• Regulated Medical Device: Any device that meets the definition of a medical device as defined in
section 201(h) of the FD&C Act, and that has been cleared or approved by the FDA review of a
premarket submission or is otherwise classified by the FDA.
NOTE: The 21st Century Cures Act (12/13/2016)amended the definition of “device” in the Food, Drug
and Cosmetic Act to exclude certain software functions, including some described in this guidance
document. FDA continues to assess emerging technology and provides guidance to represent their
current thinking on this topic. For additional information, refer to
https://www.fda.gov/medical-
devices/software-medical-device-samd/your-clinical-decision-support-software-it-medical-device.
PROCEDURE
The study team should first clarify if the app/software used is collecting/storing individually identifiable
health information (IIHI), private health information (PHI), or sensitive information in the protocol,

Table of Contents
Page 188 of 273
protocol addendum (if a multisite study) and consent. If not, then OIT review is not needed, and the
study may proceed with the IRB review. Review our information chart
, to make sure the app/software
needs to go to OIT for review.
If the study is not conducted at Emory, see the following:
• For studies taking place at CHOA, ask the team to submit a request to CHOA (via CHOA intranet-
Careforce) using https://choa.careforceconnection.org/docs/DOC-37465
. To access information on
how to submit for security review, refer all questions to [email protected].
• For studies taking place at Grady: email the device and data use the information to the Grady
privacy officer, D’Andrea Morning, at djmornin[email protected]
for her review and approval. Upload
the email in the study history for our records.
If OIT REVIEW IS NEEDED
If the mobile device/app is collecting/storing IIHI, PHI or sensitive information (from now on, “needs OIT
review), direct the team to put an IT ticket through this link:
https://emory.service-
now.com/sp?id=kb_article&sys_id=e5c2cd88f5cdf1c055c77bd8604896c2 The form has the information
to provide under “more information”
MOBILE MEDICAL APPS USED IN RESEARCH STUDIES
1. Determine whether the mobile platform+app is a medical device per FDA definition (aka intended
for use in the diagnosis of a disease or other conditions, or in the cure, mitigation, treatment, or
prevention of disease; see References and Definitions above)
a. If not, the mobile device/app does not need to be included in the device section of the
Smartform. Skip to section 2 to review ownership of the mobile device.
2. Check to see that the device is listed in the protocol and consent form:
a. If the protocol is sponsor generated, the “supplement to sponsor” protocol template should
provide the relevant information.
b. Check to see that the device is listed in the Device Section of the Smartform.
c. Review whether the device is likely to be considered a non-significant risk device, significant
risk device, or does it fall under the category of devices for which the FDA will practice
enforcement discretion.
d. For studies with enforcement discretion, where the mobile app is considered a device but
the FDA will not require an IDE submission despite not being previously approved (see
FDA
guidance) On page 12 of this guidance, you can find some examples where the FDA intends
to exercise enforcement discretion because the medical app poses a low risk to patients.
• The study team should select the following options when filling out the device
section of the Smartform:
o Under question 1 (“Select each device…”), they should add the name of the
device (if not found on the list) and attach a manual for a device. If there is
not device manual, PI should provide clarification for why there is not a
device manual. “This is not an FDA approved use of the device” should be
selected under Q2.0

Table of Contents
Page 189 of 273
o Under question 2 (“Device exemptions…”), they should select “Claim of
abbreviated IDE…).
o Under question 3, they should attach their completed
Mobile Medical apps
worksheet
3. Once screening procedures are complete, send to FB or expedited reviewer, as applicable.
• Mobile medical apps under enforcement discretion will need a device risk determination. The
risk determination can be made by the expedited reviewer.
• For studies with mobile medical devices, except if they fall under FDA enforcement discretion,
the study will have to go for a FB device determination unless:
i. the device is already FDA approved per indication OR
ii. The device is IDE exempt
4. PROCEED TO “COMMON PROCEDURES” SECTION BELOW.
MOBILE PLATFORMS IN A RESEARCH STUDY (NOT MOBILE MEDICAL APP)
1. Before ensuring the mobile platform needs OIT review, determine the ownership/possession of the
mobile device/app (whether a medical device or not). This should be described in the protocol.
2. Confirm with the study team whether the mobile device will be given to the subject or if the subject
will be using their own mobile device.
a. If the subjects will be using their own mobile device, have study teams confirm the following:
(the following should be addressed in the protocol and in the consent)
• Will an app need to be downloaded onto their own mobile device?
• Will the information on the app be encrypted?
• Will the study team be able to monitor the activity of the mobile device? If so, how will this
be done?
• What are the security measures being taken to ensure the confidentiality of their
information?
• How will the app be removed from their device?
o The removal of an app from a subject’s device should be included and detailed in the
“exit” procedures associated with a subject’s completion of the study.
i. If the subjects will be provided the mobile device, have study teams confirm the
following: (the following should be addressed in the protocol and in the consent):
• Cost questions
o Is the subject responsible for paying for the device (the device itself)?
o Who will be paying for the fees (e.g.: data, phone and texting fees)?
o If the device is being “overused” (beyond what was assumed to be
needed for the purposes of the study) who pays for the additional fees
• Misuse and accidents
o What will the study team do if the mobile device is lost or stolen? Are
subjects liable for damages?
o Is there a security measure put in place to deactivate or wipe the mobile
device remotely? If so, what is it? Under what circumstances will this
occur?

Table of Contents
Page 190 of 273
o What will be done if the mobile device is misused (e.g. visiting illicit
sites, using the device for personal reasons such as texting or calling
friends).
o If the mobile device is used for personal unauthorized reasons, who will
be responsible for the ensuing fees? What are the consequences?
• Access:
o Will the study team be able to monitor the activity of the mobile
device? If so, how will this be done?
2. PROCEED TO “COMMON PROCEDURES” SECTION BELOW.
COMMON PROCEDURES FOR ALL OF THE ABOVE
5. Determine security of data and PHI:
• Will the study team be gathering identifiable data (i.e. location) from the device? If so, why and
how will it be used?
• The study team must provide details of how information gather via the mobile device will be
stored and how it will be protected
• The study team should establish a system or schedule of contacting subjects via the device (if
this is part of the study), so as to avoid calling in the presence of third parties or in situations
where it is dangerous to answer (e.g. while driving).
Additional Resources:
Examples of Feedback from FDA
Bypass procedures
OIT vetted options and process guidance
OIT report review
After the OIT review occurred, the OIT security rep will send a report to the study team and the IRB.
The OIT report will be emailed to our listserv. The listserver will upload the report to the study
submission as a comment to study staff. Alternatively, the study team may provide a copy of the report.
The report will contain information about the findings. See the following chart to ask for additional
information from the team:
Risk level definitions, mitigation timelines, and risk acceptance criteria

Table of Contents
Page 191 of 273
Note: The risk level may be noted as "Info". These can be treated as “low” as defined above.
If a finding was deemed more than low, we should expect a letter from the study team department
Director/Chair (or VP/Dean as applicable) to allow the use of a device/software.
References
• Emory IRB P&Ps: Chapter 65- INVESTIGATIONAL MEDICAL DEVICES
Risk Level
Description
Mitigation Timeline
Risk Acceptance Criteria
Critical
The Security Review has
determined that the current
level of risk associated with the
finding is critical (severe) in its
current state.
The risk must be fully
remediated or mitigated to an
acceptable level within 30
days if system is live. If the
system is not yet live, the risk
must be fully remediated or
mitigated to an acceptable
level before the system goes
live or is connected to a
production Emory network.
Critical risks can only be
accepted by VP/Dean level
leadership with the consent of
Emory’s Chief Information
Security Officer.
High
The Security Review has
determined that the current
level of risk associated with the
finding is high (substantial) in
its current state.
The risk must be fully
remediated or mitigated to an
acceptable level within 60
days if the system is live. If
the system is not yet live, the
risk must be fully remediated
or mitigated to an acceptable
level before the system goes
live or is connected to a
production Emory network.
High risks can be accepted by
Director/Chair level
leadership. VP/Dean level
leadership must be informed
that the risk is being accepted
by the study team department
Director/Chair.
Medium
The Security Review has
determined that the current
level of risk associated with the
finding is medium (moderate)
in its current state.
The risk must be fully
remediated or mitigated to an
acceptable level within 90
days.
Medium risks may be
accepted by study team
department Director/Chair
level leadership.
Low
The Security Review has
determined that the current
level of risk associated with the
finding is low (tolerable) in its
current state.
Risk remediation is
recommended, but not
required at present.
Low risks do not need to be
explicitly accepted.

Table of Contents
Page 192 of 273
• FDA Guidance on Mobile Medical Applications at https://www.fda.gov/media/80958/download
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
4/20/2017
Updated broken links
1/31/2018
Added information about Grady Privacy officer
2/7/2018
Clarifying that mobile medical apps under enforcement discretion do not have to go to FB for risk
determination.
5/17/2018
Removed need to email Derek or Maria; added information from CHOA IT person and the link to our
information chart
7/25/2018
Added that we should let OSP/OTT (add email OTT) know if the study is under a security review when the
study is funded. Added information about LITS security review report.
4/14/2020
Updated contact for CHOA for security reviews
5/18/2020
Overall update to align with the use in the new system.
5/24/2021
Updating link for CHOA security review
2/9/2024
Update to OIT reference and links

Table of Contents
Page 193 of 273
SOP Title:
Certificate of Confidentiality Process for non-federally funded studies
SOP Category:
Study Management
Established:
6/26/2012
Last Revision:
5/5/2021
DEFINITIONS
Certificate of Confidentiality: The CoC protects the identifiable research records from forced disclosure,
such as subpoena. The CoC only covers information in the research study records, and research results
placed BY THE STUDY into the medical record. If there is documentation elsewhere of the subject’s
stigmatizing condition (e.g. HIV diagnosis at a non-Emory clinic, or mention of illicit drug use already in
their EHC or other medical record from self-report at a past physical exam), the CoC will NOT protect
that.
DEFINITIONS
Certificate of Confidentiality: The CoC protects the identifiable research records from forced disclosure,
such as subpoena. The CoC only covers information in the research study records, and research results
placed BY THE STUDY into the medical record. If there is documentation elsewhere of the subject’s
stigmatizing condition (e.g. HIV diagnosis at a non-Emory clinic, or mention of illicit drug use already in
their EHC or other medical record from self-report at a past physical exam), the CoC will NOT protect
that.
PURPOSE
In response to the 21
st
Century Cures Act, federal agencies have started granting Certificates of
Confidentiality (CoC’s) to more research projects.
NIH: The NIH now automatically grants Certificates of Confidentiality (CoCs) for all NIH- funded human
research studies, effective, October 17, 2017. They will still grant CoC’s to non-NIH-funded studies upon
request if certain criteria are met related to the sensitivity of information.
Per NIH, sensitive information includes (but is not limited to) information relating to sexual attitudes,
preferences, or practices; information relating to the use of alcohol, drugs, or other addictive products;
information pertaining to illegal conduct; information that, if released, might be damaging to an
individual's financial standing, employability, or reputation within the community or might lead to social
stigmatization or discrimination; information pertaining to an individual's psychological well-being or
mental health; and genetic information or tissue samples.
For NIH- funded studies that automatically receive a CoC: please ensure that the template CoC
language is in the ICF and check the box for “Certificate of Confidentiality” in the pre-review form.
CDC: The CDC will automatically grant a CoC, without requiring a separate application, if the IRB
determines that the information is sensitive. The study team must inform their CDC funding contact of
the IRB’s determination.
FDA: The Cures Act revisions made issuance of a CoC mandatory for federally funded researchers
“engaged in biomedical, behavioral, clinical, or other research, in which identifiable, sensitive
information is collected (including research on mental health and research on the use and effect of
alcohol and other psychoactive drugs. For non-federally funded research, FDA has been issuing

Table of Contents
Page 194 of 273
discretionary CoCs pursuant to the amended statutory requirements, on a case-by-case basis upon
application to FDA since enactment of the Cures Act. To ensure that discretionary CoCs are issued to
those entities who can comply with the requirements of the statutory provision, it is recommended that
only sponsors or sponsor-investigators, submit requests for discretionary CoCs. This will help eliminate
duplicative request to FDA for the same human subject research.
Non-NIH, Non-CDC funded studies: CoC’s may be requested proactively by the study team, or they may
be a requirement of the IRB during initial review.
The remainder of this SOP applies only to non-NIH-funded studies obtaining sensitive information
from subjects.
SCOPE
The SOP applies to all non-NIH-funded human subject research where the research activities are likely to
solicit information considered to be sensitive per the NIH definition above. See our IRB website here for
information about CoCs: http://irb.emory.edu/forms/coc.html
and the NIH’s information here:
https://era.nih.gov/files/Cert_Confidentiality_Ext_userguide.pdf
RESPONSIBILITIES
• IRB Study Analyst – Owner of the study to which the CoC applies or will apply; can also acknowledge
own Modifications that simply upload approved CoCs; delays stamping of consent forms until CoC is
in place, if applicable
• Institutional Official (I.O.) –Completes the Assurance Statement and e-signs electronic CoC
applications before they are sent to NIH.
• IRB Reviewer (contingency or expedited) – May require CoC for new studies, or ongoing studies
when more sensitive information starts being collected or identifiers are introduced; Determines if
subjects can be enrolled prior to CoC being in place
PROCEDURE
Processing studies that will have a CoC (new studies or Modifications)

Table of Contents
Page 195 of 273
Charted Procedure:
Written Procedure:
1. If any study documents reflect that there is a CoC in effect for the study, or that one will be applied
for, OR the IRB requires a CoC to minimize risk to subjects:
New Study/ Mod received by
analyst. CoC document uploaded
in Local Site Documents or Study
Related Documents?
Yes.
Continue Processing the study/MOD
until approval, then process CoC
application per chart 2 below.
No.
Give the team two options:
Move toward approval w/o CoC to enroll
earlier.
-Team submits 2 ICF versions (1 with CoC
language and 1 without)
*it will be up to a reviewer/FB to determine if
this is appropriate for the study
Final outcome: approved.
-Write approval letter and state that
consent form version (insert version
with CoC language) may not be used
until a Modification is submitted with
the approved CoC.
-Stamp consents w/o CoC language for
immediate use and process CoC
application per chart 2 below.
MOD with CoC in Local Site
Documents section is submitted
-Analyst processes the memo as per
chart 2 instructions but can
administratively approve the Mod and
stamp the CoC language ICF
*be sure to remove the non-CoC
language ICF from the documents tab!
Wait for CoC before enrolling.
-Keep CoC language in ICF and move forward with
review.
-Write approval letter and state that enrollment
cannot begin until CoC in place. Do not stamp
consents.
-After approval, continue processing CoC application
per chart 2 below.
Final outcome: team needs CoC before
enrolling.
-The team can remove the ICF without
CoC language and send changes back.
-Continue processing. Write approval
letter and state that enrollment cannot
begin until CoC in place. Do not stamp
consents.
-After approval, continue processing
per chart 2 below.

Table of Contents
Page 196 of 273
a. Ensure that the consent form language is consistent with the presence of a CoC. (Must have the
required language describing the CoC, and also NOT have language about records being subject
to subpoena [since CoC prevents disclosure based on subpoena]); require changes if necessary)
i. If no, request that the study team make the required changes.
b. Determine if the study has already obtained a CoC.
i. If study team will be applying for CoC, there are three choices depending on whether study
team needs IRB approval to obtain funding or to start preliminary activities:
(i) Leave IRB approval pending until CoC is in place: acceptable if the study team does not
need to do any human subjects research activity or obtain a grant award until then (CoC
approval from NIH can take some time). No enrollment until CoC is in place: study team
submits only the CoC-version of the consent form; or
(ii) Issue IRB approval without stamping consent forms until a Modification is submitted
with the approved CoC; or
(iii) Study team can request enrollment of subjects prior to the approval of the CoC:
1. Study team submits two versions of the ICF(s) – with and without CoC language (NIH
advises against using language like “we plan to obtain a CoC”).
2. For full board studies, put a note in the Full Board Information form asking IRB to
determine if study team can start enrolling prior to CoC being approved, despite
potential exposure of sensitive data to subpoena.
3. For expedited studies, include note to reviewer asking them to make the
determination listed above (and email to follow up if they neglect to do so)
4. If IRB does not decide to allow enrollment pre-CoC, the no-CoC version must be
removed as a contingency for approval.
2. Post-approval:
a. If Option (i) or (iii) above: Proceed as with any study.
b. If Option (ii) above, CoC version(s) of ICFs must NOT BE STAMPED until the CoC is approved by
the NIH.
i. In approval letter (whether full board or expedited) state that the consent form version
[insert version with CoC language] may not be used until a Modification is submitted with
the approved CoC.
ii. Once Modification is submitted to upload the approved CoC in “Local Site Documents”
section of smartform, IRB Study Analyst may “approve” (really just acknowledge) the
Modification.
iii. If study used Option (iii) above, Modification must remove non-CoC versions of consent
form, so they will no longer appear as “Finalized.”
Handling CoC applications

Table of Contents
Page 197 of 273
Note: if this is a new multi-site study for which a CoC already exists (due to other sites already enrolling
with a CoC in place), please skip to the CoC Assurance section.
Chart 2
1) Study team completes the electronic Certificate of Confidentiality (CoC) Request form.
2) Institutional Official receives "Verification and submission of CoC Application” email notification
from the NIH's Electronic Research Administration system and forwards to Emory IRB Listserv where
it is directed to IRB analyst that owns the study.
a) IRB Analyst checks eIRB SaaS to confirm Project Description entered by team aligns with
applicable study, consent includes CoC language and that the study has been approved by the
IRB.
b) Analyst confirms application questions #13, #14, & #15 on CoC application have Emory
Institutional Official's (IO’s) name, email and phone number. If not, study team may need to
start over if they cannot edit the application and resend.
c) IRB Analyst forwards verified CoC Application email to IO re: CoC to confirm that the application
is ready for his sign-off.
d) Institutional Official completes the Assurance Statement in the electronic Certificate of
Confidentiality Request form and clicks the "Submit" button.
3) Study team/PI & Institutional Official receive "CoC Application has been approved" email
Study Team completes electronic CoC
request application and includes
Institutional Official's name in #13.
Institutional Official receives
"Verification and submission of CoC
Application email notification from the
NIH's Electronic Research
Administration system, and forwards
to Emory IRB Listserv where it is
directed to IRB analyst that owns the
study.
Analyst checks eIRB SaaS to confirm
Project Description in CoC application
aligns with applicable study, consent
includes CoC language, and that the
study has been approved by the IRB.
Analyst confirms application questions
#13, #14, & #15 on CoC application
have Emory Institutional Official's(IO)
name, email and phone number (if
not, study team may need to start
over if they cannot edit that)
IRB Analyst forwards verified CoC
Application email to IO re: CoC to
confirm that the application is ready
for his sign-off
Institutional Official completes the
Assurance Statement in the electronic
Certificate of Confidentiality Request
form and clicks the "Submit" button.
Study team/PI & Institutional Official
receive "CoC Application has been
approved" email
If Study has already been approved
with CoC language in the consent form
by the IRB, the Study team/PI should
create and submit a Modification and
upload the final Certificate of
Confidentiality in the Local Site
Documents section of eIRB SaaS
smartform.
IRB Analyst will approve the CoC
Modification to acknowledge receipt
and finalize the ICF documents that
contain CoC language.

Table of Contents
Page 198 of 273
If Study has already been approved with CoC language in the consent form by the IRB, the
Study team/PI should create and submit a Modification and upload the final Certificate of
Confidentiality in the Local Site Documents section of eIRB SaaS smartform.
4) IRB Analyst will approve the CoC Modification to acknowledge receipt and finalize the ICF
documents that contain CoC language.
There is guidance for researchers available on the web. Please direct researchers there if they have
questions, http://www.irb.emory.edu/forms/coc.html
.
Handling CoC Site Assurances:
Assurance documents are signed when Emory will invoke a CoC already in place in a multisite study,
where Emory is not the main study site. The process for CoC assurances is basically the same as the
above procedure. The only difference is that since the CoC is already in place, the team will submit a
statement of assurance (on their departmental letterhead) rather than an application. There is template
language available in the CoC Kiosk if the study team needs it. In the assurance process, the “packet” will
include the cover memo, assurance document, approved informed consent form, and approval letter.
The folder name to be saved on the H drive should be titled “CoC Assurance XXXXX PI name.”
Background:
https://grants.nih.gov/policy/humansubjects/coc/what-is.htm
https://grants.nih.gov/policy/humansubjects/coc/who-can.htm
https://grants.nih.gov/policy/humansubjects/coc.htm
https://www.fda.gov/media/132966/download
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/13/2015
Clarification of process
12/23/2015
Revamping of SOP following current NIH requirements.
3/1/2016
Language addition to reflect use of IRB subcommittee use, use of department letterhead and
signature of PI for requests, as well as other language clarifications without content changes.
11/22/2016
Updated name of template Memo to IO
5/5/2017
Addition of section: handling Coc assurances; other minor clarifications
11/1/2017
Added charts, clarified SOP for new NIH CoC policy from 10/1/2017, other minor clarifications.
1/18/2019
Updated Dr. Wynes with Dr. Sherer
7/15/2020
Updates to the process
5/5/2021
Updated to follow new Online Certificate of Confidentiality System User Guide dated 06/25/2020.

Table of Contents
Page 199 of 273
SOP Title: Data sharing certifications including genomic data sharing
SOP Category:
Study Management
Established:
10/31/2018
Last Revision:
10/31/2018
PURPOSE
The purpose of this document is to explain the steps to follow if reviewing a study under a data sharing
requirement, including genomic data sharing.
DEFINITIONS
• Genomic data sharing repository (e.g. dbGap): public repository for individual-level phenotype,
exposure, genotype, and sequence data, and the associations between them. dbGaP assigns stable,
unique identifiers to studies and subsets of information from those studies, including documents,
individual phenotypic variables, tables of trait data, sets of genotype data, computed phenotype-
genotype associations and groups of study subjects who have given similar consents for use of their
data.
• Institutional Certifications: Institutions are responsible for assuring, through an Institutional
Certification, that plans for the submission of large-scale human genomic data to the NIH meet the
expectations of the Genomic Data Sharing Policy
(examples of research within the scope of the GDS
Policy can be found in the Supplemental Information to the Policy). An Institutional Certification
must accompany the submission of all large-scale human data to the NIH Database of Genotypes
and Phenotypes (dbGaP). The Institutional Certification (for sharing human data), should also be
provided to the funding NIH Institute or Center prior to award, along with any other Just in Time
information (for extramural researchers) or at the time of scientific review (for intramural
researchers).
• Provisional Institutional Certification: to be used in a situation such as for a prospective study where
the IRB has not completed its review of the protocol and therefore the institution cannot attest to
all of the elements of the formal Institutional Certification
RESPONSIBILITIES
• IRB analyst – reviews checklist submitted by study team and verified if consent has required
information to allow data sharing
• IRB Director- reviews information
We have a guidance for investigators in our website at
http://www.irb.emory.edu/forms/Data_Sharing.html
Study teams should fill these forms as appropriate:
Institutional Certification Request form for Emory submitting data
Institutional Certification Request form for Emory not submitting data
PROCEDURE
• If this is a urgent, very tight-turnaround request, strongly recommend to OSP analyst that OSP
instead sign the “Provisional Institutional Certification
” – state that IRB believes this is an
appropriate use of the Provisional version. This form can be found under the NIH Institutional
Certifications
page.
• Request comes into IRB from OSP and/or Study team
• Refer study team to IRB form to fill out and await it

Table of Contents
Page 200 of 273
• The IRB analyst reviews as follows:
o Does the Consent Describe Sharing? Y/N
If sharing is described if it is optional? Y/N or N/A
o Genetic or Genomic Research Described? Y/N
o If Genetic or Genomic Research Described is it optional? Y/N or N/A
o Data Use Specifications:
o Appropriate for DbGaP submission (if applies): Y/N
o Unrestricted or restricted areas: Y/N
o Controlled access: Y/N
o If study in question was approved by the IRB before January 23, 2015, ICF will only need to
make reference to sharing data or samples, but not as explicit as above
o If the study wants to access a database or repository approved after January 23, 2015, a
waiver of consent will not be valid for this purpose.
• After this process was completed, forward the information the study team sent and what was
reviewed to the IRB Director or designee.
• The IRB Director or designee will determine if this information is consistent with the approved study
and protocol, and forward letter to IRB chair for signature
o If the IRB Director or designee finds that the study did not allow for this use, she will
communicate with study team
• After the Chair signs the letter, the IRB Director will forward to IRB analyst to communicate with
study team and to log a comment in eIRB with the letter.
REFERENCES
• NIH webpage: Institutional Certifications.
• dbGAP submission process: chart
• NIH Guidance: Expectations for Non-NIH-funded Submission Requests
• NIH Institutes and Centers Genomic Program Administrators
• Provisional Institutional Certification form
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 201 of 273
SOP Title:
COI: Handling Studies with Study Team Conflict of Interest
SOP Category:
Study Management
Established:
4/27/2015
Last Revision:
08/15/2024
PURPOSE
The purpose of this document is to detail the steps necessary for the IRB to review studies with an
investigator conflict of interest.
SCOPE
The SOP applies to all human subject research where the COI Office / Investigator has identified that the
investigator has a conflict of interest.
RESPONSIBILITIES
• COI Office- identifies and reviews potential conflicts of interest, creates COI management plans
when needed
• IRB Study Analyst- processes studies in which study team members have disclosed a COI in eIRB;
manages the COI ancillary review
• AVP of the HRPP or delegate: Notifies email of their IRB-approved management plans and deadlines
thereof; also for following up if investigators do not comply by the deadline, by requiring the
submission of reportable new information (RNI) submission and further action if needed.
• IRB CoRe– IRB subcommittee responsible for reviewing and approving (with additions as needed)
COI management plans that affect human subjects research approved by the Emory IRB
• IRB Full Committee- responsible for re-reviewing management plan when the investigator objects to
any significant additional requirements recommended by IRB CoRe.
PROCEDURE
NOTE: The changes to P&Ps that allow for the CoRe review described below went into effect at the P&P
subcommittee meeting on March 17, 2015
COI and External IRB Studies
• If a study team member has or discloses a COI for a study going to be reviewed by and External IRB:
o If this is a NEW industry sponsored study, it should be reviewed by our IRB unless single IRB
review is required by funding agency or OHRP.
o If this is a NEW study mandated to be reviewed by a single IRB or otherwise needs to go to an
external IRB:
Trigger COI Ancillary Review in XIRB submission
Await the release of the Emory COI office’s management plan before issuing
institutional sign-off
Provide the COI management plan to the reviewing IRB for their review (the
plan document includes suggested informed consent language)
Ensure that financial disclosure language, if required, is present in the Emory
informed consent form before moving study to “Active” state
o If this is an ONGOING study reviewed by an external IRB, that previously had no COI:
IRB Director will receive notification that there is a COI, along with the
management plan

Table of Contents
Page 202 of 273
IRB Director will email the study team (conflicted party and PI), copying the
Emory IRB Reliance Team, asking them to submit the management plan to the
reviewing IRB within 10 business days
IRB Director and Reliance Team should set a calendar reminder for 10 business
days, to confirm the study team has complied
If study team has not complied, IRB Director emails a reminder, and copies COI
office.
If after another 10 business days the study team has still not complied, IRB
Director will inform: the Reviewing IRB (providing the management plan) and
the Department Chair
IRB Study Analyst
1) New study or MOD submission indicate that the PI or a study team member have a COI (for
disclosures that come initially from COI office, please see (3) below)
a. Ask the study team if they have disclosed to the COI office (unless this has been done, based on
the content of the comment), and if so, what the status of COI review is. If this takes place
outside of eIRB, document the exchange in the History.
b. Contact Team Q, copying AVP, informing them of the COI disclosure and the IRB study number,
and what is known of COI review status.
c. Add the COI office as an ancillary review, if the disclosure is new or pending.
d. Await feedback as you continue processing the submission as follows:
i. For Full Board study/AM, the item can be assigned to an agenda, as long as COI remains a
pending issue until there is confirmation of COI Committee review or finding of no conflict;
and until a management plan (if applicable) is accepted by the investigator and the plan is
approved (with additions if needed) by IRB CoRe. Include this requirement in the Agenda
Item Notes and add a logged comment in the History.
ii. For expedited items, do not send to the reviewer until COI review is complete, and if
applicable, a management plan is accepted by the investigator, and the plan is approved
(with additions if needed) by IRB CoRe.
1. Upon receipt of an IRB CoRe-approved management plan (as an email and/or logged
comment), the IRB Study Analyst should review the management plan against the eIRB
record to ensure that the study team has made the required changes if any (e.g. revising
the protocol, Smartform (e.g. recruitment or consent sections, and including updating
the COI section, if necessary), consent form(s), etc). Log comment in History stating that
you have verified this.
2. After confirming that the revisions have been completed, the IRB Study Analyst should
send the item for the appropriate further review (may be full board initial review,
contingency review if COI was a pending issue, or expedited initial review). Indicate in
your pre-review notes (if sending for initial full board review) that COI management plan
has been reviewed and implemented by the IRB CoRe.
2) Continuing Review
a. If question 5 of the CR application has been checked, verify if this is being disclosed for the
first time.
i. If already disclosed, there is no need to take further action regarding COI (unless you do
not see evidence that there was a COI review or a management plan, in which case
contact the COI specialist and IRB Director).

Table of Contents
Page 203 of 273
b. If the disclosure is new, ask the study team if they have submitted it to the COI office (unless
this has been done).
i. If far in advance of expiration, send back to the study team to get their response;
otherwise can obtain a response via call/email/logged comment as needed, but
document the exchange in study History.
c. Contact Team Q, copying AVP, informing them of disclosure and study number, and what is
known of COI review status. Log a comment in the History stating that you have done this.
d. Add the COI as an ancillary review for the CR.
e. Process the CR as usual; this is not a pending issue (though a Modification may need to be
submitted once a management plan is approved, per section (3) below).
f. Alternatively, if the conflict was incorrectly identified, you may need to ask the study team
to correct and send back.
3) COI Management Plans for Ongoing Studies In Absence of eIRB Disclosure
Sometimes a COI will develop in an ongoing study, and the study team does not initially submit a
Modification with that information in eIRB. Therefore you may hear about it first from the COI
Office t, as follows:
a. COI staff member will send an email and log a private comment to the analyst and IRB
Director, informing them that the study team has been given 10 business days to submit a
Modification to implement the COI management plan.
b. IRB Study Analyst should set a calendar reminder for 10 business days to double-check that
the study team complies with updating the study to implement the COI management plan
and check the COI box.
c. If no Modification is submitted within the timeframe, log a comment to the study team
reminding them. Also, email the IRB Director and Team Q for escalation, and log a comment
in the History stating that you have done this.
i. If there is a pending Continuing Review, make that PENDING until the study
team has complied with the management plan
d. Handoff responsibility for monitoring management plan completion to the Team Q and IRB
Director. Director will escalate to COI Office and Department Chair if needed.
COI COI staff member
1) Notification of Investigator or Study COI is sent to you by IRB Analyst, due to disclosure in eIRB by
COI staff member:
a. The COI staff member will reply to the analyst to acknowledge receipt of the disclosure, and will
provide an estimated turnaround for next steps (if not able to complete remaining steps
immediately)
i. If the COI Committee has already determined a management plan, the COI staff member
will log a comment in eIRB to notify the IRB analyst of the current approval status
ii. If the COI management plan has not been determined by the COI Committee, the COI staff
member will log a comment with that information.
iii. If the COI Committee determined there was no conflict or plan needed, the COI staff
member will complete the ancillary review accordingly.
b. The COI specialist will follow steps below once COI Committee review is complete (Step 2 when
COI is present; Step 3 when COI Office review finds no COI exists)
2) COI Exists, Management Plan is finalized by COI Committee and Dept/VP Review Period has Elapsed

Table of Contents
Page 204 of 273
a. The COI specialist saves the Management Plan on IRB shared drive (:\General\COI\COI Mgmt
Plans, under the appropriate folder).
b. If study already is present in eIRB:
i. Email the management plan and study link to non-clinical IRB CoRe with the following
information using the “CoRE COI determination needed” email template
(H:\General\COI\COI Omnibus Forms and Determination Letter Templates):
1. If an investigator’s COI involves multiple studies, you may include the multiple
management plans in one email – see template – summarizing them separately if there
are differences in the plans. The result is still multiple omnibus forms however.
ii. Set reminder to follow up with CoRE to ensure votes received in timely manner.
iii. After a determination is made by IRB CoRe (with at least 3 agreements, including a Vice
Chair), update the COI Omnibus form and save on H:Drive (H:\General\COI\COI Mgmt Plans)
after creating a folder. Also save PDF of all CoRe email responses/votes.
a. If multiple studies were involved in the above CoRe email, you may use one folder to
hold all documents for those studies. Include the eIRB numbers and PI names in the
name of the folder.
iv. Create Determination Letter – one per management plan (even if CoRe reviewed multiple
plans at once);
1. If the Management Plan is accepted as is:
a. Use the Accepted As Is COI Mgmt Plan Determination Letter, found at
“H:\General\COI\COI Omnibus Forms and Determination Letter Templates” to craft
a final determination letter. Save in case folder on shared drive.
2. If there are changes to the Management Plan (e.g. adding additional requirement):
a. Use the Not Accepted COI Mgmt Plan Determination Letter, found at
“H:\General\COI\COI Omnibus Forms and Determination Letter Templates” to craft
a final determination letter. Save in case folder on shared drive.
3. Forward to IRB Director for final approval of language and authorization for digital
signature, then create PDF of signed letter and save on H: drive folder.
v. Update COI Office records and tracking spreadsheet accordingly
vi. Email approved management plan and the signed PDF’d letter to IRB Director, copying IRB
analyst.
1. Include links to each study involved and who is analyst (on each, if more than one
study); otherwise not clear
2. If study already approved and ongoing, include in the email a deadline of 10 business
days to implement, and reminder to the IRB Analyst to check in 10 business days.
“Analyst(s): Please set Outlook reminder to verify that changes are made within 10
business days; if not, escalate to Team Q and Director.”
3. If study is not yet finally approved, indicate to IRB Analyst that the COI management
plan implementation must be a pending issue.
vii. Upload approved management plan and letter into eIRB submission’s History as Private IRB
Comment (not visible to study team); also paste in text of above email.
viii. Log Comment to Study Team (visible to study team) stating simply that a Conflict of Interest
Management Plan has been approved by the IRB and the IRB Director will be emailing
shortly with the formal letter and further instructions.
c. If study not yet present in eIRB
i. Record case on COI tracking spreadsheet and await inquiry from IRB staff once study is
submitted.

Table of Contents
Page 205 of 273
3) COI Office finds No COI Exists
a. No required action
Escalation Procedure when Study Team Does Not Comply with Mgt Plan Within Timeframe
1. If changes to the consent documents are needed, the modification should be submitting within 10
business days of the notification to the study team. If the modification is not submitted during this
time, refer the issue to Director for follow up
2. The IRB Director will communicate with the study team, letting them know they are now required to
submit an RNI due to the delay.
IRB Director:
• As a member of CoRe, submit opinions re: COI management plans on a timely basis (14 calendar day
total turnaround for CoRe determination)
• When COI Specialist sends IRB CoRe determination letters, email those directly, within 2 business
days, to conflicted party and PI stating that the IRB has accepted the management plan (as-is or with
required changes) and that either (a) the requirements of the plan have already been met in the IRB
submission and no further action is needed, or (b) the required changes must be submitted in eIRB
(1) before the IRB can issue final approval, or (2) via Modification within 10 calendar days, and if the
Modification is not submitted within that timeframe a Reportable New Information submission may
be required.
o Copy IRBA and COI Program Specialist
o Update COI tracking spreadsheet when email sent
TARGET TURNAROUND TIMES
Time 0 (Zero) = When COI Office receives notification from the conflicted party that they have accepted
the COI management plan, or when the timeframe for objection has elapsed
Targets
When the plan is approved by CoRe “as is”:
• 14 calendar days (two weeks) from Time 0 to when IRB CoRe determination letter uploaded in
eIRB and emailed to IRB analyst and Director
• Two business days for Director to send the plan to conflicted party and PI
• Ten calendar days for the study team to make necessary changes in eIRB via a MOD, before
referred to Team Q
When CoRe requires changes:
• 14 days from Time 0 to when IRB determination letter is uploaded into eIRB and emailed to IRB
analyst and Director
• 2 business days for Director to send required changes to the conflicted party via email
• 5 business days to allow the conflicted party to object (must be stated in an email to PI)
• 10 additional calendar days for a final CoRe determination letter to be prepared by COI Program
Specialist, sent to IRB analyst and Director, and Director to send to conflicted party and PI
LOG OF SIGNIFICANT CHANGES

Table of Contents
Page 206 of 273
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/7/2015
Replaced the SOP titled “Conflict of Interest Management Plan Process” with all new procedures
8/17/2015
Added to IRB COI Liaison Analyst’s responsibility to keep IRB analyst informed of plan status.
5/18/2016
Updating the number and composition of CoRE needed for acceptance. Also updated responsibilities
between IRB and COI.
12/20/2016
Major overhaul of entire SOP. Removal of IRB COI Liaison role.
6/14/2020
Revisions to SOP to improve understanding and readability.
10/23/2020
Updated/enhanced SOP section for studies reviewed by External IRB. Removed name of VPRA
(Sherer).
08/15/2024
Updated responsibilities

Table of Contents
Page 207 of 273
SOP Title:
Institutional Conflict of Interest
SOP Category:
Study Management
Established:
3/21/2016
Last Revision:
3/21/2016
BACKGROUND
This SOP outlines the Emory IRB’s responsibilities pursuant to the Emory Institutional Financial Interests
Involving Human Subjects Research (Policy 7.24, http://policies.emory.edu/7.24
DEFINITIONS
• Institutional Financial Conflict of Interest involving Human Subject Research exists when the
University Institutional Conflict of Interest Review Committee determines that a Significant
Institutional Financial Interest held by Emory University or an Institutional Leader can
significantly and directly affect or reasonably appear to affect the institutional processes for the
design, conduct, reporting, review, or oversight of human subject research
• Licensed Intellectual Property in Human Subject Research: When Emory licenses its intellectual
property (IP), the University may receive equity in a company as a result of a licensing
agreement for Emory IP; receive royalties or other fees as compensation for the use of that IP;
and/or may receive equity or other financial interest as part of a co-investment in a licensee or
related company.
• Gifts: In compliance with the Emory Gift Acceptance Policies & Procedures, Emory Policy 3.7,
any gifts to Emory must be unconditional, in furtherance of Emory’s charitable mission, and non-
reciprocal. Per the procedures identified in Emory Policy 3.7, any gifts of equity in individual
companies will be sold as soon as it can be practically and legally accomplished. These
procedures will be used for any gifts of equity in companies that utilize Emory Intellectual
Property to produce a drug, device, diagnostic, etc. that is involved in Human Subject Research
at Emory
• Significant Financial Interests of Institutional Leaders: Pursuant to this policy, Institutional
Leaders shall recuse themselves from any business decision, allocation of University resources
or personnel, approval process, or oversight review process involving a company with which
they have a Significant Institutional Financial Interest that is related to Human Subject Research
at Emory. If recusal is not possible in order to carry out their University obligations, they must
divest the Institutional Financial Interest. Any exception must be reviewed by the Provost and
the Vice President for Research Administration, who may request an advisory opinion from the
University Institutional Conflict of Interest Review Committee. For example, a Department Chair
should not review or approve an IRB protocol for Human Subject Research when she has a
Significant Institutional Financial Interest in the sponsor or provider of test material in the
protocol. Emory may receive gifts from corporate donors that may also sponsor research
involving human subjects. When a gift meets the Institutional Financial Interest threshold, the
gift must be reviewed pursuant to the procedure in Policy 7.24, Section III, E.
ROLES AND RESPONSIBILITIES
• Investigator: To the best of their knowledge, Investigators shall identify the use of Emory
Intellectual Property in Human Subject Research on the IRB application. Those protocols shall be
forwarded to the Vice President for Research Administration or his designee for assessment and
review.

Table of Contents
Page 208 of 273
• Office of Technology Transfer: shall compile a list that includes: (i) all entities in which the
University holds an equity interest as part of a licensing arrangement; and (ii) a list of
technologies sorted by licensee where Emory has received more than $100,000 in royalties
annually These lists shall be updated every six months or as requested by the COI Review Office.
Using this list of entities, the COI Review Office will search the IRB databases to determine
whether an identified entity is the financial supporter for the study. The COI Review Office shall
refer any identified IRB protocols and licensing information to the Vice President for Research
Administration. The Vice President for Research Administration, or designee, shall follow the
procedures for assessment and review.
• The Office of the Vice President for Health Affairs Development (for Gifts to Emory): shall
provide to the IRB Department a list of corporate donors that give more than $500,000 in cash
per annum to a Department or Center. On a reasonable basis, the IRB will review the list of
donors against a list of active protocols. If a listed Donor is the sponsor or financial supporter of
Human Subject Research, the IRB will refer the research and gift proposal for review by the Vice
President for Research Administration, or his designee. The Vice President for Research
Administration, or designee, shall follow the procedures for assessment and review.
• University Conflict of Interest Review Committee: When an Investigator submits an explanation
of Compelling Circumstances the VPRA may send the protocol to an ad hoc independent
organization, including an external IRB independent of Emory, or may form an ad hoc
committee that includes one member from outside Emory. These experts will formulate a
recommendation as to whether an Institutional Financial Conflict of Interest exists and how it
could be managed. In reviewing the research, the Significant Institutional Financial Interest, and
the explanation of Compelling Circumstances, the Committee will use the Criteria for Evaluating
an Institutional Financial Conflict of Interest in Policy 7.24, Section III, E.3.b.1. The Committee
may determine that the Significant Institutional Financial Interest is too great and the research
should not occur at Emory, unless divestiture is possible prior to the commencement of the
study.
• Vice President of Research Administration (VPRA): Receives notification when Institutional
Conflict of Interest is revealed in the context of a human subjects research protocol. The VPRA
shall make an initial assessment of whether a Significant Institutional Financial Interest related
to the research exists, then require either disclosure in publications/consent/presentations, or
ask investigator for compelling circumstances why the research should proceed at Emory.
• IRB Director: Ensures that IRB-COI Liaison follows up as required when alerted of ICOI disclosure
• IRB-COI Liaison: Communicates with COI Office re: any institutional COI issues disclosed to either
IRB or to COI Office
• IRB Analyst: Alerts IRB-COI Liaison and IRB Director if any studies come to Analyst where
Institutional COI is checked.
PROCEDURE
Procedures For When Human Subjects Research Involves Emory Licensed Intellectual Property, or is
Funded by Entities Who Provided Large Gifts to Emory
1. The IRB is alerted to the presence of Institutional COI by either
a. the investigator (in the eIRB application)
i. IRB Analyst must alert the IRB-COI Liaison and the IRB Director

Table of Contents
Page 209 of 273
ii. The above individuals will inquire with the Emory COI office and the VPRA to see
what stage Emory’s review of the potential ICOI is in
iii. If/when the VPRA has determined that there is a significant financial institutional
COI, and he or the ad hoc Committee has agreed that the research may still be
conducted at Emory (i.e. the conflict can be adequately managed), he will share the
management plan with the IRB, which may include things like disclosure in the
informed consent form, and/or referring the study to an external IRB for review
1. If external IRB will review and study not yet approved at Emory,
communicate to study that outside IRB submission is required, and refer to
Team Theta for any additional assistance needed.
2. If study will remain at Emory IRB (rare), transfer ownership of study to IRB-
COI Liaison.
b. Or the VPRA
i. Will then skip to step (a) (ii) above
2. The IRB of record (either Emory IRB or external IRB such as WIRB) must review the Institutional
Conflict of Interest management plan and determine if any additional elements are needed.
NOTE: The IRB shall review the management plan as part of its initial review of a new protocol. If an ICOI
arises after initial IRB approval has been granted, the IRB will review the management plan following
notice by the UCOIRC
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 210 of 273
SOP Title: Cost Option for Clinical Trial Agreements and ICFs
SOP Category:
Study Management
Established:
9/20/12
Last Revision
6/13/2022
PURPOSE
The purpose of this SOP is to outline the procedure the IRB analyst uses to determine the correct cost
option language to include in informed consent forms.
SCOPE
The SOP applies to research studies that have informed consent forms.
PROCEDURE
1. Review the response to question #2 in the “Clinical Research/Expanded Access only” section of the
smart form to determine if the study has billable items or services.
2. If the study has billable items or services, the appropriate cost language in the consent form is
option #2. The Office of Clinical Research (OCR ) will not provide documentation of this selection
and the IRB will assume this is the default selection.
3. For studies with no billable items or services, the correct cost option is option #1.
4. If a study team has inserted option #3, they must provide documentation of the cost option from
OCR. This justification should be routed to a TL or Director prior to assigning for review to
determine if there is an ethical concern regarding participants paying for research-related
procedures.
If the OCR pre-award staff identifies a cost option other than #2 for a clinical study, they will send
notification of the correct option to the IRB listserv. The notification will include the IRB study ID.
The IRB Listserv attendant will forward this email to the designated person who will look up the
study and upload the document as an attachment with a comment like: “CTA Cost Option is #___
per attached.”
NOTE: If the consent form has been approved with the incorrect language, a modification will be
required.
In this scenario, the IRB Analyst posts a comment in the History and/or Agenda Item Notes about the
Cost Option being verified and correct in the ICF (or not yet verified, etc.)
FOR PEDS/CHOA STUDIES
1. OCR does not provide cost options to IRB for studies done by Pediatrics.
a. In some cases OCR may send a preliminary cost option to the Peds RAS, who then needs
to finalize the option based on further review.
2. Peds RAS may get input from CHOA, if there are CHOA procedures. This process is conducted
between these offices and we do not need to get input from CHOA directly.
3. Peds RAS will email our listserv with the Cost option again.

Table of Contents
Page 211 of 273
a. If confirmation of the cost option is not received, please email Kim Caroline
)
Note: the Monday report to the IRB listserv is about the In Case of Injury option, not the Cost Option.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/6/2017
Addition of CHOA process
5/21/2021
Modified PED/CHOA studies procedure
3/28/2022
Updated the Cost option process
6/13/2022
Minor formatting updates, rewording for clarity

Table of Contents
Page 212 of 273
SOP Title: Sensitive Study Status
SOP Category:
Study Management
Established:
9/23/2015
Last Revision:
08/11/2023
PURPOSE
The purpose of this document is to outline the procedure to process sensitive study requests/
determinations.
SCOPE
The SOP applies to clinical research studies in which the study team seeks to keep the subject’s
consent/HIPAA and other sensitive information from being uploaded into EeMR due to the stigmatizing
nature of the information.
BACKGROUND
In order to ensure that EHC physicians are aware of a patient’s participation in a clinical trial, and how
that participation might affect their symptoms or treatment options, the Emory Office of Quality
requires that study information be placed in any EHC medical record that a study participant may have.
If a subject has an EeMR Record, or gets one created when they come seeking medical treatment at
EHC, then those documents are automatically transferred into the subject’s EHC EeMR.
For studies with stigmatizing information that would not already be part of their EHC medical record,
the study team can request Sensitive Study Status from the IRB, and complete the “Clinical Research Key
Points for Sensitive Study” document to keep certain study-related information out of the EeMR. In
either case, the consent form must accurately reflect what study information will be placed in the EHC
EeMR.
PROCEDURE
• New Studies/Modifications
1. Study team should request “Sensitive Study Status” by selecting yes to Q5.0 under the For
Clinical Research/Expanded Access Only of the Smartform.
2. IRB Analyst should review the consent form to make sure that the appropriate template
language regarding the exclusion of the consent form and study information from the
medical record is included.
3. IRB Analyst should send the study for appropriate review (FB or expedited). (if declined by
expedited reviewer and study team disagrees with outcome, request should be referred to
FB).
4. IRB Analyst should include the reviewer’s determination of sensitive study status in the
approval letter, if granted (if declined, and study team agrees with outcome, then consent
should be reverted to non-sensitive template language)
5. For studies with sensitive study status, the IRB Analyst should forward the approval letter to
.

Table of Contents
Page 213 of 273
• Misc.
a. For approved studies that are found to contain Sensitive Study Status language in the
consent, but have not been granted sensitive study status (i.e. IRB review overlooked this
discrepancy):
1. Consult with the IRB Director and/or OCR to check to see if the consents are actually
being uploaded to ERMS
2. If consents are being uploaded, have study team submit an modification and RE to
correct the discrepancy.
3. If they are not being uploaded, then have study team submit an amendment to use
simple language re: no study information will be placed in any EHC medical record you
might have, instead of the sensitive study template language.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/11/2020
Update to align with new system
8/11/2023
Removed reference to CRKP form in study smart form

Table of Contents
Page 214 of 273
SOP Title:
Imaging Studies
SOP Category:
Study Management
Established:
3/10/2014
Last Revision:
6/8/2018
PURPOSE
The purpose of this SOP is to detail to IRB Analysts what are the required elements for processing
imaging studies.
SCOPE
This SOP applies to any study that utilizes any of the below referenced imaging modalities as a research
intervention.
DEFINITIONS:
• Emory University Radiation Safety Committee (RSC): The RSC reviews Research that involves the
use of radioactive isotopes, x-rays or other radioactive materials. Requirements for when RSC
review of a Research protocol is necessary can be found at the following website:
http://www.ehso.emory.edu/documents/guideline-for-rsc-review-of-human-research-
studies.pdf
• Radioactive tracer: A radioactive molecule that can be sent through the body's circulatory or
urinary system, with its progress followed by a radiation-sensitive machine.
• Radioactive contrast: a solution or colloid containing radioactive material used for visualizing
soft tissue structures. Such contrast media indicate their positions or distribution in the body by
their gamma ray emissions.
PROCEDURES:
Below are two categories of modals related to imaging studies. For additional assistance please see
guidance on referring to Radiation Safety.
fMRI/MRI scans
• Determine if the study could be expeditable vs Full Board.
o In general, determine if fMRI or MRI being used does not utilize any radioactive tracers
(some MRI’s use these). If they do, please make sure the study team registers with RSC
and the study is assigned to a full board meeting.
o fMRI/MRI studies may be expedited if no contrast is being used and the overall study is
minimal risk and all other procedures fit into the expedited categories. The designated
reviewer must be a medical doctor.
• If the study is using a radioactive tracer or contrast, make sure that the risks section of the consent,
contains the following information:
o A warning that the subject should not participate if they have any type of metallic object
implanted in their body. The MRI may cause these objects to move or heat up.
o The loudness of the machine (subjects are usually given earplugs)

Table of Contents
Page 215 of 273
o The machine requires the subject to be in an enclosed space for a prolonged duration of
time. If the subject is claustrophobic they may wish to opt out of the study. (language may
be altered for open MRI)
• The incidental findings language from our Modular Consent Language document (posted on IRB
website) must be included verbatim in every study that utilizes an fMRI or MRI for research
purposes only, in either the risk section or its own section called “Incidental Findings”
Please note
• A clinical trial with research scans added that are not done as standard of care may need to add
incidental findings language to the consent form.
• Often times these studies are done through the Psychology department. The Psychology
department is not part of the covered entity and may not need a HIPAA Authorization form.
However, if the study staff includes investigators (doctors) that are part of a covered entity, and the
study involves billing and treatment, then HIPAA may apply.
• Many of the fMRI and MRI studies will come from Dr. Gregory Berns laboratory called the Facility for
Education and Research in Neuroscience (FERN). Although the following fMRI/MRI directions were
specifically designed for studies submitted by the Berns lab they are applicable to all studies using
fMRI’s and MRI’s as a research intervention.
Other Scans
For studies utilizing scans such as MUGA, Bone Scan, PET, PET/CT, Myocardial Perfusion, VQ Scan,
Thyroid, or exposure to radiation of any kind, please make sure the study team lists “radiation” in the
“Biomedical Research” section, to trigger Radiation Safety Committee review, and the study is assigned
to a full board meeting.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
11/6/2014
Expanded the scope from just fMRI and MRI to cover all studies where imaging is done for research
2/18/2015
Adding information about other imaging that may require referral to RSC and review by FB
4/27/15
Adding note about the potential for HIPAA requirement based on study staff.
12/10/2015
Administrative changes made for clarity and note about clinical trials with research scans.
6/8/2018
Minor clarifications

Table of Contents
Page 216 of 273
SOP Title:
How to Handle GWAS Data Use Certification Requests
SOP Category:
Study Management
Established:
12/08/ 2009
Last Revision:
9/16/2016
SUMMARY
Genome-Wide Association Studies (GWAS) and “large-scale genomic analyses” are increasingly
common. Investigators at Emory may wish to participate in GWAS via NIH-supported repositories by
submitting genetic samples/data and/or requesting the use of it. As part of the process for submitting
genetic samples/data, the investigators need to have a Data Use Certification signed by an appropriate
Emory official. Per the latest NIH genomic data sharing policy effective January 25, 2015, additional
steps are needed for NIH-funded studies including large scale genomic analyses. The IRB has a role in
certifying/approving data sharing plans at the time of grant submission for these types of studies, as
well as when the data is ready to be submitted to a public repository.
If requests come to the IRB either from OSP or the investigators to certify/sign-off on data sharing plans
or data sharing activities, the IRB must have the following:
1. The letter or form that we are being asked to sign, pre-filled with information about the
study and the data sharing restrictions as proposed by the investigator
2. The grant application including the data sharing plan portion
3. Indication of whether the samples from which the data was gathered were collected
prior to, or after, January 25, 2015 (or both)
4. Copies of all informed consent forms used to collect the samples from which the data
was gathered.
The Emory IRB office handles these requests and arranges for signature by the IRB Director after
verification that all criteria are met. In her absence, a Co-Chair, Vice Chair, or an Associate or Assistant
Director may sign after verifying in the protocol record that all criteria are met.
IRB Staff: if you have questions about GWAS or genomic data sharing issues, please check with IRB
Director.
REFERENCES
• Learn more in the NIH GWAS Points to consider at:
http://gds.nih.gov/pdf/PTC_for_IRBs_and_Institutions.pdf
• You may also obtain more information at http://gds.nih.gov/
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
9/16/2016
Updated with information about 1/25/15 policy change

Table of Contents
Page 217 of 273
SOP Title: Humanitarian Device Exemption (HDE) Studies
SOP Category:
Study Management
Established:
8/8/2013
Last Revision:
11/3/2020
PURPOSE
The purpose of this document is to describe the IRB process for reviewing studies under a Humanitarian
Device Exemption.
SCOPE OF SOP
The SOP will apply to Emory human subject research working under a HDE.
DEFINITIONS
• Investigational Medical Device: means a device, including a transitional device that is the object of
an investigation. An investigational device is permitted by the FDA to be tested in humans but not
yet determined to be safe and effective for a particular use in the general population and not yet
licensed for marketing.
• Humanitarian Use Device (HUD): medical device intended to benefit patients in the treatment or
diagnosis of a disease or condition that affects or is manifested in fewer than 4,000 individuals in the
United States per year – 21 CFR 814.3(n). To obtain approval for an HUD, a humanitarian device
exemption (HDE) application is submitted to the FDA.
• Humanitarian Device Exemptions (HDE): allows use of an HUD; the HDE application is similar in both
form and content to a premarket approval (PMA) application, but is exempt from the effectiveness
requirements of a PMA. An HDE application is not required to contain the results of scientifically
valid clinical investigations demonstrating that the device is effective for its intended purpose. The
application, however, must contain sufficient information for FDA to determine that the device does
not pose an unreasonable or significant risk of illness or injury, and that the probable benefit to
health outweighs the risk of injury or illness from its use, taking into account the probable risks and
benefits of currently available devices or alternative forms of treatment. Additionally, the applicant
must demonstrate that no comparable devices are available to treat or diagnose the disease or
condition, and that they could not otherwise bring the device to market.
• Sponsor: A person who takes responsibility for and initiates a clinical investigation. The sponsor may
be an individual or pharmaceutical company, governmental agency, academic institution, private
organization, or other organization. The sponsor does not actually conduct the investigation unless
the sponsor is a sponsor-investigator
RESPONSIBILITIES
• IRB analyst- initially screens application before sending it to IRB Full Board or the IRB Chair.
• IRB Chair or Co-Chair – reviews the progress reports submitted by the sponsor or sponsor-
investigator. The Chair or Co-Chair must be a clinician
PROCEDURE
Initial Review Submission
Note: The Emory IRB cannot be the IRB of record for community physicians who want to use a HUD for non-
research purposes. We can only serve as the IRB of record for Emory physicians using HUDs at Grady or Emory. If

Table of Contents
Page 218 of 273
the HUD is being used at St. Joseph’s Hospital, CHOA, etc., their IRB (or a commercial IRB) should review the HUD
submission.
Non-research HUD
Screen the study using the HUD NS checklist. When adding this to the FB agenda, make sure to attach
the FB section to your pre-review notesThe study team should provide the following documents with
the submission (forward
this checklist to the treating physician for their information):
• A copy of the HDE approval order
• A description of the device
• The product labeling
• An ICF is not required if used under the FDA HDE-approved use, but the study team should submit
an information sheet for the patient. The information sheet should describe a general definition of
the FDA’s HDE program, a brief description of the device and related procedures, risk/benefit ratio,
and physician contact information if the patient experiences a device-related adverse event.
o If the HDE holder has developed a patient information packet, this packet always should be
distributed to patients prior to receiving their HUDs. Labeling for the HUD may also be made
available to the patient to provide further information regarding the device’s HUD status and
possible risks/benefits packet for the patient. See here
for the Emory IRB template.
• A summary of how the physician proposes to use the device, including a description of any screening
procedures, the HUD procedure, and any patient monitoring with follow-up visits, tests, or
procedures. Refer here
for a protocol outline.
• The IRB reviewer also may request that the physician submit documentation that he/she is qualified
through training and expertise to use the HUD.
The staff should confirm the approval status of the HUD. The information can be found at
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfHDE/HDEInformation.cfm
.Letter template
information can be found at H:\General\QA Working Files\Forms, templates and Guidance\Letter
templates & guidelines.
• Ancillary reviews required: Department Review, Biosafety/Radiation Safety, and Clinical Research
Key Points Summary.
• Requires Full Board initial review- most criteria in the worksheet don’t apply since the project isn’t
research
• Treating staff are not required to have CITI certification.
Research HUD
Even though HUD’s are “approved” devices, the sponsor may still want to gather data from the uses of
the device in order to support a premarket approval. If this is the case, then the PI should provide a
research protocol and research informed consent and HIPAA Authorization. The use in the study may
require an IDE. This information should be reviewed and approved by the IRB, and all regular IRB P&Ps
should apply. You should refer to the “
eIRB Processing of New Study Applications- Preliminary Analysis
through Approval” for next steps.
Note: Off-label use of an HUD for research purposes requires an IDE. And unless it is an emergency,
before an HUD is used off-label treatment, the FDA recommends that the HDE holder obtain FDA
approval of the use following the compassionate use policy for unapproved devices. IRB approval is also
required per our policies and procedures.

Table of Contents
Page 219 of 273
Continuing Review Submission
Non- research HUD
• When a continuing review application is submitted to the Emory IRB, the IRB analyst will screen the
information.
• As a part of Continuing Review, the IRB reviewer may request the HDE holder to provide safety
information on the HUD provided to the FDA in periodic reports required under 21 CFR Section
814.126(b)(1).
• It is mandatory for the PI to provide a detailed list of each use of the HUD within the previous
approval period. Summaries should include a brief description of the patient’s condition (with no
identifiers), how the device was used, whether or not an information sheet was provided to the
patient, and the patient’s outcome.
o The analyst will route the continuing review, as permitted by the FDA regulations, to the
clinician IRB Co-Chair for expedited review. The chair will make the decision if the HDE should
be reviewed at Full Board instead of expedited review for any reason.
o The IRB analyst and reviewer should compare the uses made of the device to the approved
scope of the HDE/indication.
o If there is a case where the PI appears to have used the device off-label, without informing the
IRB in advance or within 5 days of the use, the analyst in charge of the continuing review should
alert the Q team of possible non-compliance (NC).
Reportable new information submission for NC/UPs and Emergency/Compassionate Use
• Research or non-research HUD
o The study team should follow Emory IRB’s usual reporting P&P for reporting protocol
deviations, noncompliance, and unanticipated problems.
o If the protocol deviation involves an emergency use or compassionate use in a single
patient:
If the device user is or can be added to the HUD submission, study team should
submit an RNI to report this matter. Team Q will send the event to FB using the
appropriate emergency use omnibus form.
If the device user is not and cannot be on the HUD submission (per the PI choice),
the device user must submit a new eIRB application of their own. Please make
Team Q aware so they may follow up and take ownership of that submission.
A study team can add the new device user to an approved study to avoid a new
submission.
Note: The Emory IRB will not review HUD compassionate/emergency use request for community doctors. If the
original HUD PI wants to add this community physician to the submission, this may be allowed. Please contact the
IRB Director for more information about this process.
Close-Outs
• If the treating physician has use the device in new patients since the last CR approval (to the close-
out), he/she should include a detailed list of each use of the HUD within this period. Summaries
should include a brief description of the patient’s condition (with no identifiers), how the device was
used, and the patient’s outcome. The close out should be reviewed by a medical vice-chair.
• If the close-out is submitted and the treating physician has not used the device in new patients since
the last CR, the close-out can be processed by an analyst.

Table of Contents
Page 220 of 273
REFERENCES
• 21 CFR Part 812 (investigational devices)
• 21 CFR Part 814 (premarket approval of medical devices)
• 21 CFR Part 860 (device classification procedures);
• 21 CFR Parts 862 –892 (device type classifications)
• IRB policies and procedures
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
1/5/2015
Replaced “should” with “must” to state Chair/Co-Chair must be a clinician; added HUD definition
5/14/2015
Added New Study and Reportable new information submission sections.
6/29/2015
Added close out section and information for template letter.
9/16/2016
Added links to current tools and clarified Emory IRB position on reviewing HUDs for community
physicians.
6/14/2020
General updates to clarify process, formatting corrections. Added a link to the HUD checklist.
11/3/2020
Added additional required steps in the review process

Table of Contents
Page 221 of 273
SOP Title:
ResearchMatch.org as a recruitment tool
SOP
Study Management
Established:
7/28/2014
Last Revision:
5/14/2015
PURPOSE
The purpose of this document is detail the steps necessary to add ResearchMatch.org as a recruitment
tool.
SCOPE
The SOP applies to all studies that want to add ResearchMatch.org (RM) as a recruitment tool.
PROCEDURE
• Emory researcher includes RM template language (
ResearchMatch IRB Protocol Template
Language) in the research protocol or, more likely, as an addendum (separate document) to the
protocol, uploaded in the “Research Design” section next to the protocol. Alternatively, it can be
uploaded as a stand-alone document, along with #2 below, to the “Recruitment and Payment”
section of the IRB application form in the question where flyers are uploaded.
• Study team adds the use of RM as a recruitment document in the “Recruitment and Payment”
section of the eIRB and adds the RM template language as a stand-alone document (usually the
same text as the main recruitment flyer, but with all research contact information removed).
• Emory IRB reviews and approves the submission in eIRB
• Study team follows the ResearchMatch Registration Instructions
to register in RM. A copy of
IRB’s approval of the overall study, as well as the text for the recruitment ad, is required. When
RM use is being added via Modification, registration with RM should be done after the
Modification is approved by the IRB.
• Emory IRB ResearchMatch Liaison receives email from RM.org, alerting them that there is a
study pending approval. Emory IRB RM Liaison logs into RM system (IRBEmoryRM/ IRB2010RM),
confirms study has IRB approval to use RM as recruitment tool with correct ad content, and sets
study expiration date in RM.org.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
10/22/2015
Addition of links for template language and instructions. Modification of Process
flow

Table of Contents
Page 222 of 273
SOP Title:
Checking Biosafety Approval Status
SOP Category:
Study Management
Established:
5/2/2012
Last Revision:
5/11/2016
PURPOSE
The purpose of this document is to describe the procedures to check the status of Biosafety Approval.
SCOPE
The SOP applies to all studies that may require Biosafety approval (ex. all recombinant DNA use in
humans directly (i.e vaccine) or indirectly (autologous cells sent off for treatment with lentiviral vector
and then infused back to patient)).
RESPONSIBILITIES
• Biosafety Analyst – review and update the Emory Bio_Chem Protocol Box. In addition, Biosafety
analysts may log a comment in eIRB regarding the status of the Biosafety review.
• IRB Analyst – ensure that all studies needing Biosafety review have obtained the necessary approval.
• IRB Superuser – may be required to provide Biosafety approval in eIRB
• Study team – select Biosafety review as a necessary review in the eIRB Smartform and apply for the
necessary approval.
PROCEDURE
Once the study team has selected Biosafety as a necessary ancillary review, the IRB analyst should wait
for confirmation of the completion of the Biosafety review by either:
1) Waiting for a Biosafety analyst to log an approval comment into the study OR
2) Review the Biosafety Emory Box
File (Bio_Chem Protocols)
a. Note, only select IRB individuals may have access to this Emory Box file. If you do not have
access, you can ask either an Associate or Assistant Director/ Biosafety analyst to provide you
access.
If you find that one of your studies has been approved by Biosafety, you can ask a superuser to do the
approval in eIRB; likewise if you find out it’s not actually required for a study, a superuser can remove
the requirement in eIRB.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/11/16
Updating SOP to reflect Biosafety Committee use of Emory Box

Table of Contents
Page 223 of 273
SOP Title:
Processing Studies that will use Deception or Incomplete Disclosure
SOP Category:
Study Management
Established:
5/17/2018
Last Revision:
5/17/2018
PURPOSE
The purpose of this document is to outline special considerations when processing studies that will
utilize deception or incomplete disclosure. The IRB will determine when certain restrictions apply, and
consider the extent to which the deception in a given study interferes with the subject's ability to give
informed consent. The IRB will need to distinguishing whether "deception" or only "incomplete
disclosure" (without deception) is involved, whether there is sufficient justification for use of such
measures, and whether there is an appropriate consent and debriefing process in place.
SCOPE
The SOP applies to any study that will utilize deception or incomplete disclosure.
RESPONSIBILITIES
• IRB analyst – Review studies to determine whether deception or incomplete disclosure is included in
the research plan.
• IRB Associate or Assistant Director and/or Director- Serve a resource for analysts in cases where
appropriate process of review in unclear.
DEFINITIONS
Deception-occurs when an investigator gives false information to subjects or intentionally misleads
them about some key aspect of the research. Examples include:
• The subject is given a "cover story" which falsely describes the purpose of the study, but
provides a feasible account of the researcher's objective.
• Participants complete a quiz and are falsely told that they did poorly, regardless of their
performance.
• Participants who don’t know they are in a research study are observed to see how they behave
when they find valuables (e.g., wallet, laptop) unattended in a public location.
• The study includes a researcher's "confederate," an individual who poses as a participant, but
whose behavior in the study is actually part of the researcher's experimental design.
Incomplete disclosure -occurs when an investigator withholds information about the specific purpose,
nature, or other aspect of the research. Withholding information may or may not be considered
deception. Examples include:
• Participants are asked to take a quiz for research, but they are not told the research question
involves how background noise affects their ability to concentrate.
• Participants are told they are completing a survey to evaluate customer service when the true
purpose of the study is to correlate psychological responses with patient care satisfaction.
Incomplete disclosure that is also deception. An example:
• The study involves audiotaping or videotaping of subjects without their knowledge or prior
consent.
PROCEDURE
Considerations when triaging studies:

Table of Contents
Page 224 of 273
In keeping with federal regulations and ethical codes established by the Belmont Report and the
American Psychological Association, the IRB will consider the following criteria when reviewing research
involving the use of deception or incomplete disclosure:
• The study must not involve any more than minimal risk to the subjects.
• The use of deceptive techniques must be justified by the study’s prospective value AND there
should be no reasonable alternative method that would be equally effective (i.e., the researcher
must demonstrate that the deception is necessary to conduct the study).
• Prospective subjects must not be deceived about any physical or psychological risks,
discomforts, or unpleasant emotional experiences of the study.
• If the study design allows, subjects should be told during the original consent process that some
information is being withheld or is incomplete, and that they will receive more information after
the research is over. This is sometimes known as “authorized deception” because it provides
participants with an opportunity to decide whether or not to participate, knowing that they
aren’t receiving complete information. However, researchers often believe that even vague
references to hidden purposes will affect subjects' behavior and make the study impracticable.
Investigators should either add such language to their consent forms when it is possible or note
in their protocols why it is not feasible to do so.
• In addition, the research must meet the criteria for a waiver of one or more elements of
informed consent.
• Whenever appropriate, researchers should debrief participants. The debriefing should occur as
early in the study as the design permits, preferably at the conclusion of a subject’s participation,
but no later than the conclusion of the research.
Please note: Research involving incomplete disclosure but no deception may be reviewed as
Exempt. Research employing deception may not be reviewed as Exempt under the Pre-2018
Common Rule. Under the Post-2018 Common Rule, such research may be reviewed as Exempt
if it otherwise meets the Exemption criteria and the deception is authorized (see [§ 46.104
(d)(3)(iii)]).
Research that involves incomplete disclosure, or that involves mild deception where the topic is
not sensitive and the participants are not vulnerable, may be reviewed as Expedited with the
discretion of the designated reviewer. Research that involves deception where the topic may be
sensitive and/or the participants may be vulnerable should be referred to full board for review,
in parallel with consultation with the Committee C Chair in case she determines that the study
may receive expedited review. The IRB Analyst should also consult with the Associate or
Assistant Director or IRB Director to determine whether Full Board review is appropriate.
PROCESS FLOW
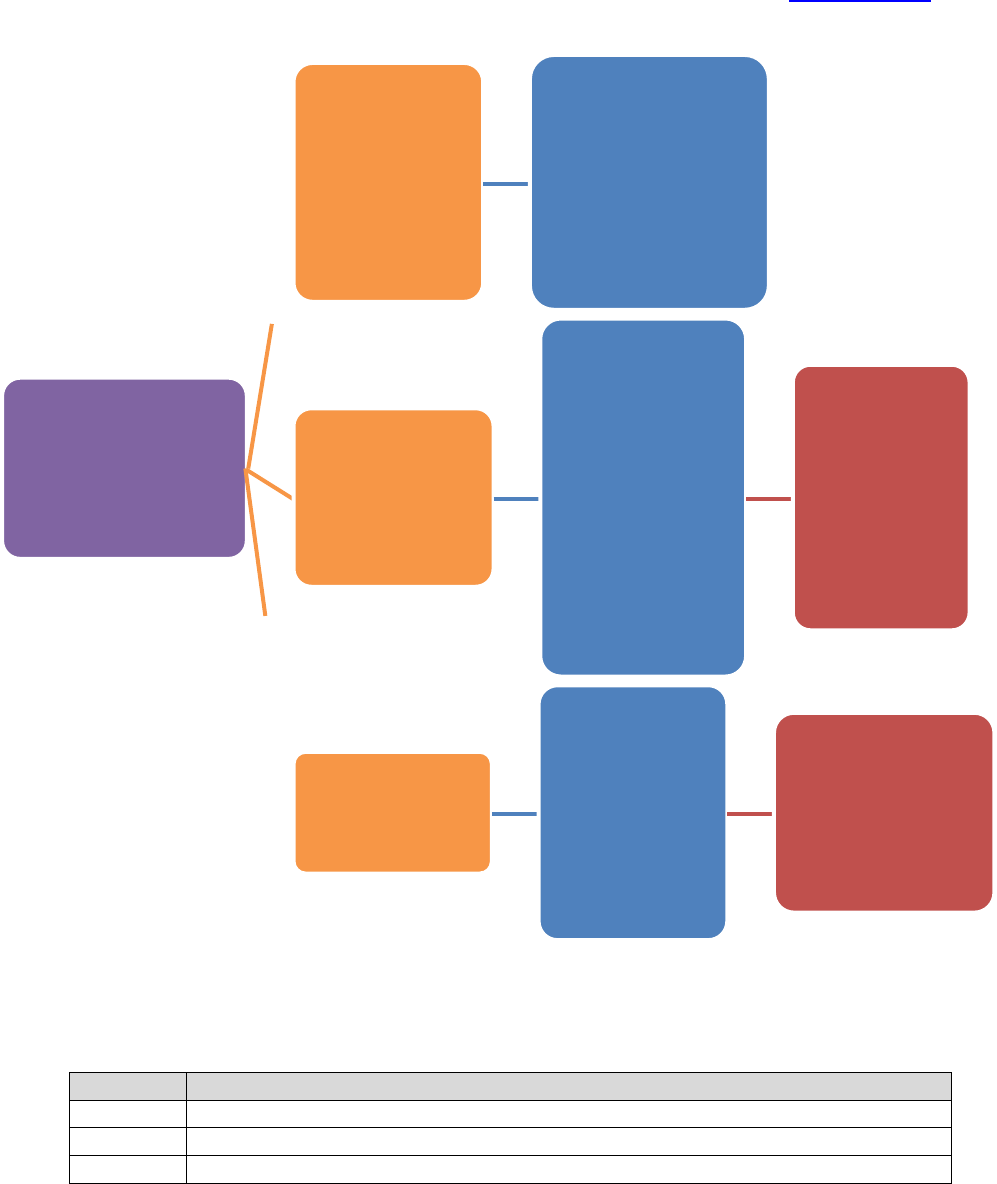
Table of Contents
Page 225 of 273
*Adapted from University of North Dakota IRB’s ‘Guidance on the Use of Deception or Incomplete Disclosure in Research”
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
Determine whether
study involves
deception and/or
incomplete
disclosure
Incomplete
Disclosure?
If study meets the
criteria for Exempt
review, may be an
option. If Exempt
categories do not
apply, may go
Expedited.
Incomplete
Disclosure,
Mild Deception,
or both?
If mild
deception &
topic is not
sensitive or
participants are
not vulnerable,
analyst should
consult with the
Committee C
Chair or
Director
If possible,
expedite. If
not
expeditable,
assign to
CMTE C for
review
Significant
Deception?
Analyst should
consult with
the
Committee C
Chair or
Director.
Not Expeditable?
Assign to CMTE C
for review

Table of Contents
Page 226 of 273
SOP Title:
St. Joseph and John’s Creek study site process
SOP Category:
Study Management
Established:
11/27/2013
Last Revision:
1/31/2018
PURPOSE
The purpose of this document is detailing the steps for handling a study submission including or adding
John’s Creek and/or St. Joseph (SJHC) as a study site in order to ensure compliance with institutional
requirements and ethical and religious directives (ERDs). Per an MOU (created when EHC, St. Joseph’s,
and Johns Creek became partially owned by a Joint Operating Company [JOC]), Emory IRB is the IRB of
record for any studies involving Emory or Emory Healthcare personnel, that have St. Joseph’s or Johns
Creek as study sites. EHC has set up an Emory Clinic (TEC) location in leased space on the St. Joseph’s
campus; research studies might take place there, or at the actual hospitals, or both. St. Joseph’s does
not have an IRB or Research Oversight Committee anymore. Rebecca Heitkam will review protocols and
consents to verify compliance with ERDs.
SCOPE
The SOP applies to all new studies and Modifications including St. Joseph and/ or John’s Creek as a study
site (other than chart reviews and clinical trials/research conducted at other Emory sites (e.g. EUH) but
isolated diagnostic services performed at ESJH for research purposes due to patient convenience or
equipment availability).
DEFINITIONS
• ERDs - Ethical and Religious Directives
for Catholic Health Care Services
• SJHC - St. Joseph Healthcare
PROCEDURE
IRB Analyst:
1) Review the eIRB submission overall for mention of St. Joseph and/ or John’s Creek as a location
where study activities will take place.
a. Confirm that Emory St. Joseph/Emory Johns Creek is selected as a study site in the Smartform
b. Leased space (i.e. TEC) should be reflected using the “other” site selection as well (See appendix
1 for an example)
i. Confirm with study team whether activities will take place in leased space or not.
c. Advise the team (when requesting changes) that they will need at least one physician on the study
team is credentialed to practice at SJHC (or Johns Creek, as applicable).
2) Determine if SJHC local context (ERD’s and credentialing) review is required. If not, the rest of this
SOP does not apply
a. Chart reviews do not require ERDs review.
b. Clinical trials/research conducted at other Emory sites (e.g. EUH) but isolated diagnostic services
performed at ESJH for research purposes due to patient convenience or equipment availability
do not require ERDs review, though the informed consent should include information about
where study procedures will or may take place.
c. All other studies taking place at SJHC, TEC at SJHC or Emory Johns Creek require ERD
review
3) Email the Rebecca
Heitkam (rebecca.heitkam@emoryhealthcare.org) a copy of the study protocol
and consent forms. (See appendix 2 for an example)
Table of Contents
Page 227 of 273
a. For an expedited study, email when the study is assigned to you
b. For a full board study, email once the study is assigned to a committee. Include the meeting
date and a reminder that there still may be changes required by the IRB.
4) Confirm that the study used the proper consent form template if St. Joseph/ Johns Creek are
engaged.
a. Studies taking place only at the St. Joseph’s leased space (TEC) do not need a St. Joseph’s
template, as St. Joseph’s is not considered to be “engaged” in that scenario. Note: ERD review is
still required, therefore pregnancy prevention language needs to align with their requirements.
b. Ensure that the correct terminology for “birth control” is used, per our template, and that no
specific birth control methods other than abstinence are listed (e.g. no mention of birth control
pills, condoms, etc).
5) If SJHC local context approval is not in place (per logged comment from study team) prior to IRB
approval of the study or Modification, process IRB approval as usual but add caveat to approval
letter stating “Note: No research-related activities may begin at the [St. Joseph’s/Johns Creek] site(s)
until the site ERD approval is received from Rebecca Heitkam or her designee. Any changes required
by the site must be submitted to the Emory IRB via an (other) Modification.
6) Once ERDs approval is received, the analyst will log the information as a comment to study team.
PROCESS FLOW
Appendix 1
Study Sites
• A site is where recruitment will occur
• A site is where the research will take place
• A site is data collection will take place
•
A site is where data analysis will take place
Study team submits
study, or Modendment
adding site
IRB analyst sends study
information to Rebecca
Heitkam, copying study
team.
Rebecca Heitkam
provides approval /
revisions if they review
prior to IRB review
IRB analyst submits
study/Modification to FB
or designated reviewer
IRB analyst issues final
approval with caveat in
approval letter (if site
ERD approval not
already present)

Table of Contents
Page 228 of 273
•
A site is where data will be stored
1.0 *
Indicate all locations where the Emory Investigator will conduct this study:
Emory Children's Center
Emory Clinic
Emory St. Joseph's Hospital
Emory University Hospital Clinical Research Network
Winship Cancer Institute
Other - In Atlanta metropolitan area
2.0
If "Other - ", list all other locations where the Emory Investigator will conduct the
study:
Name of the Site
IRB of record for this site
TEC at St. Joseph
Appendix 2
For expedited studies:
Hello Ms. Heitkam,
IRB0000XXX, a new study/Modification, has listed St. Joseph/John’s Creek as a study
site. Please advise the study team when your approval is granted, replying all to this
email
For Full Board Studies:
Hello Ms. Heitkam,
IRB0000XXX has listed St. Joseph/John’s Creek as a study site. The study is currently
assigned to a committee meeting on X/X/201X. Please note that the committee may
request changes to the study. Please advise the study team when your approval is
granted, replying all to this email.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
10/14/2021
Added clarification on language requirements even in TEC context & archived log of updates older
than 1 year

Table of Contents
Page 229 of 273
SOP Title: REMS study review
SOP Category:
Study Management
Established:
1/31/2018
Last Revision:
4/11/2019
PURPOSE
The purpose of this document is to explain the process an IRB analyst will follow when reviewing a study
under Approved Risk Evaluation and Mitigation Strategies (REMS) requirements.
SCOPE
The SOP applies to studies either reviewed by the Emory IRB or at Emory reviewed by an external IRB
under REMS requirements.
RESPONSIBILITIES
• IRB analyst – reviews protocol to identify if drug or biologic is under REMS program & confirms ODP
review
• IRB Associate or Assistant Director or Director- initial review of protocol to identify if drug or biologic
is under REMS program & flags study as one under REMS
• Research Integrity and Compliance Designated Person (ODP)- confirms that protocol/ICF is in
compliance with REMS requirements
PROCEDURE FOR NEW STUDIES REVIEWED BY THE EMORY IRB
1. Study will be assigned by IRB Director/designee, indicating that the study is under REMS
requirements. It is possible that an analyst will receive a study that was not flagged; in those cases,
the analyst should verify with a TL or IRB Director
2. The analyst will contact the ODP and direct the study team to fill out the REMS Investigator
Checklist.
3. The ODP will verify that the study protocol and informed consent include all the requirements for
that drug under REMS. That information can be found at this FDA website
4. When the information is complete, the ODP will log a comment under the study history indicating
that the REMS requirements are met in the submitted protocol and informed consent
5. The IRB analyst may send the study to full board (FB), before this process is completed. If the ODP
review has not been completed before the IRB meeting, the analyst should complete omnibus form
accordingly.
6. When forwarding the study for FB review, the analyst will add to the agenda items notes that the
study is under REMS requirements.
7. After the study is approved, the approval letter should include the following language:
“This study utilizes a drug(s) under FDA required REMS. You must ensure your study remains
compliant with current requirements as listed in the “REMS Document” and the
protocol/informed consent document”
PROCEDURE FOR STUDIES REVIEWED BY AN EXTERNAL IRB
• Follow same steps from 1 to 4 as above.
• The study will not be given an institutional signoff until the REMS requirements are verified.

Table of Contents
Page 230 of 273
PROCEDURE FOR MODIFICATIONS REVIEWED BY THE EMORY IRB
1. If the Mod adds a new drug under REMS requirements, follow steps as detailed before, letting the
ODP know, and making sure the ODP has reviewed the checklist. The Mod may be placed on FB
agenda prior to completion, making sure the omnibus form reflect that this process is still pending.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/21/2018
Added process for Mods
4/11/2019
Clarified that REMS process applies to research at Emory or reviewed by our IRB

Table of Contents
Page 231 of 273
SOP Title:
Prisoner Studies: Handling of New/Modification/Renewal Submissions when
Prisoners are Subjects (Application of Subpart C)
SOP Category:
Study Management
Established:
10/17/2016
Last Revision:
10/14/2021
PURPOSE
The purpose of this document is to describe the review process of new study submissions, continuing
reviews, and Modifications by the IRB analyst and prisoner representatives when research involves
Prisoners.
SCOPE
This applies to all IRB staff.
DEFINITION
Prisoner: an individual involuntarily confined or detained in a penal institution. The term is intended to
encompass individuals sentenced to such an institution under a criminal or civil statute, individuals
detained in other facilities by virtue of statutes or commitment procedures which provide alternatives to
criminal prosecution or incarceration in a penal institution, and individuals detained pending
arraignment, trial, or sentencing. Individuals are prisoners if they are in any kind of penal institution,
such as a prison, jail, or juvenile offender facility, and their ability to leave the institution is restricted.
Prisoners may be convicted felons, or may be untried persons who are detained pending judicial action,
for example, arraignment or trial (45 CFR46.303).
Important Notes
• One member of the Emory IRB shall be a Prisoner or a Prisoner Representative. A Prisoner
Representative is defined as an IRB member who has appropriate background and experience that
includes a close working knowledge, understanding and appreciation of prison conditions from the
Prisoner’s perspective.
• The fact that the Emory IRB meets the compositional requirements for the review of a Research
protocol involving Prisoners will be documented in the meeting minutes at which the Research
protocol is reviewed.
• When a prisoner representative completes a review, please contact Carol Corkran, so she can
process payments.
PROCEDURE
RESEARCH PROTOCOLS THAT INTIALLY PLAN TO INCLUDE PRISONERS AS HUMAN SUBJECTS:
Note: If a study intends to enroll both prisoners and non-prisoners, and is federally funded (thus requires
OHRP certification for involvement of prisoners), study team may be advised to consider omitting
Prisoners as a study population at initial submission, and add them via Modification after initial IRB
approval is in place, depending on how urgently they wish to begin. This would enable them to begin
enrolling non-prisoners immediately.
FEDERALY FUNDED STUDIES

Table of Contents
Page 232 of 273
If the study is federally funded, we need to obtain OHRP certification of subpart C findings before final
approval. This mean that:
• For expedited studies, you cannot send to a DR until you receive the OHRP certification
• For full board studies, you can put in a FB agenda, but the study has to be pending approval until we
receive the OHRP certification
Exempt studies:
Not Applicable - Research involving prisoners cannot be reviewed as exempt, except for research aimed
at involving a broader subject population that only incidentally includes prisoners.
Expedited new studies:
• IRB analyst should complete the top portion of the Subpart C Worksheet, and highlight the portion
to be completed by the prisoner representative. The IRB analyst should emailthe prisoner
representative to provide their comments and complete the Subpart C Worksheet, as needed. Send
the study link in the email. The prisoner rep should document their findings (if any) and upload the
worksheet via a private comment. Alternatively, the prisoner rep can email this information to the
IRB analyst. This email with the completed worksheet should be logged as a private comment in the
study history.
• After this is completed, IRB analyst should send the study to a primary designated reviewer via eIRB.
Please alert the primary designated reviewer that the application has been reviewed by the prisoner
representative, and if any changes the prisoner rep required.
• Once the primary designated reviewer has completed his/her review, and any requested changes
have been made by the study team (including the ones required by the prisoner rep), IRB analyst
should send the changes back to the prisoner rep to verify their completion. The study should be
sent to the designated reviewer so they can review the adequacy of the changes.
• IRB analyst should upload the completed Subpart C Worksheet using the “Edit Pre-review” function,
unde question 7 (add supporting documents),so it becomes part of the permanent record.
• IRB analyst documents Subpart C category in the approval letter.
• If the study is federally funded, IRB analyst must draft a letter certifying the Subpart C Category
findings to OHRP
o The letter should be drafted for the IRB Director to review (may be reviewed by Associate or
Assistant Director in Director’s absence).
o The OHRP letter will require verification of the Grant number – may be requested from the
study team if not in eIRB.
o A sample OHRP letter can be found at H:\General\Subpart C OHRP Cert
Expedited new studies not involving interactions with prisoners (i.e. involve only existing data or
record reviews):
Per OHRP, AARHPP, and Emory IRB P&Ps, review by the prisoner representative is not required for
minimal risk studies that do not involve interactions with prisoners.
Full Board new studies
• For a new study, the prisoner representative must be present, either in-person or by speakerphone,
at any meeting of the Emory IRB for FULL Board review of new research protocols involving

Table of Contents
Page 233 of 273
Prisoners and Modifications adding Prisoners. Inform the meeting Pod that a prisoner
representative is needed for the meeting.
• If a continuing review, IRB analyst must document the participation of the prisoner representative in
the review, but the prisoner representative does not have to be present for the full board review of
the application.
• If a Modification alters risks/benefits/involvement of Prisoner subjects, the information should be
reviewed at the full board. If a Modification is not altering the above, then the information can be
reviewed expedited.
• For new studies, or Modifications adding Prisoners as a subject population for the first time (see the
following section for more details on the latter case):
o IRB analyst should pre-fill the Subpart C Worksheet to be filled out at the meeting.
o After the meeting, IRB analyst should save the Subpart C Worksheet in the Research Design
section of the Smart form.
o IRB analyst needs to document Subpart C category in the approval letter. Final approval should
not be granted for federally funded Subpart C studies until we have received confirmation from
OHRP about the certification of the Subpart C findings.
o If the study is federally funded, IRB analyst must draft a prisoner certification letter to OHRP
o The letter should be drafted for the IRB Director to review (may be reviewed by Associate or
Assistant Director in Director’s absence).
o The OHRP letter will require verification of the Grant number – may be requested from study
team if not in eIRB.
o A sample OHRP letter can be found at H:\General\Subpart C OHRP Cert
Continuing Reviews:
Continuing reviews for studies that required a prisoner representative review must also be reviewed by
a prisoner representative. For full board studies, the representative must attend or call in for the review
of the renewal. Expedited renewals may use the reviewer assignment procedure listed under “Expedited
new studies” above.
Modifications:
o Any Modifications that are changes to the research require prisoner representative review if
such review was required initially.
o Any Modifications that are purely administrative and would not be considered changes to the
research requiring IRB review, per OHRP, may be reviewed without a prisoner representative’s
input.
IF A PARTICIPANT BECOMES A PRISONER WHILE ENROLLED IN A RESEARCH STUDY THAT HAS NOT
BEEN REVIEWED ACCORDING TO SUBPART C
• The study team must immediately notify the IRB (via Modification or other communication) that a
participant has become a prisoner, the IRB shall confirm that the participant meets the definition of
a “Prisoner.”
• The IRB shall ensure that either:
o The PI terminates enrollment of the subject OR

Table of Contents
Page 234 of 273
o If it is feasible for the participant to remain in the study, cease any research
activities until the IRB reviews the research under Subpart C (following the steps
outlined above) or not if it meets the “Non-DHHS” criteria per the P&Ps.
• Review the Modification and make one of the following findings:
o The study is neither DHHS-funded nor considered VA Research, and the “Non-DHHS” criteria
below are met, OR
o The research meets the criteria set forth in Subpart C of the Common Rule. The PI must provide
written documentation if he/she feels there are special circumstances that justify why research
activities should continue while the IRB reviews the research under Subpart C. The special
circumstance should be reviewed by a Vice-Chair.
Non-DHHS Criteria:
• The research is NOT conducted or funded by DHHS or Veterans Administration (VA).
• The subject was not incarcerated at the time of enrollment, and subsequent incarceration was
unexpected.
• The incarceration does not put the rights and wellbeing of the subject in jeopardy with respect to
the study.
• The prisoner representative has been consulted.
• The terms of the subject’s confinement do not inhibit the ethical conduct of the research.
• There are no other significant issues preventing the research from continuing as approved.
• This approval is limited to the individual subject and does not allow the recruitment of prisoners.
• One of the following is true:
o The subject will be at increased risk of harm if withdrawn from the research
o The research presents no more than Minimal Risk and no more than an inconvenience to the
subjects
For DHHS-Regulated Research:
The research shall be reviewed per Subpart C
• If some requirements of Subpart C cannot be met:
o If it is in the best interests of the subject to remain in the study, the subject shall remain
enrolled and the IRB shall inform OHRP of the decision along with the justification.
o Otherwise, the IRB shall advise the PI to remove the participant from the study and to
keep the participant on the study intervention under an alternate mechanism as
necessary.
• When considering whether to terminate enrollment, the IRB/PI should consider the risks associated
with termination of the subject in the study.
o If the participant cannot be terminated for health or safety reasons, the IRB should advise the
PI to keep the subject enrolled in the study and it shall review the research under Subpart C.
o If some requirements of Subpart C can’t be met, but it’s in the best interest of subject to
remain in the study, the subject shall remain enrolled and IRB shall inform OHRP of the decision
along with the justification.
o The IRB shall advise the PI to remove the participant from the study and to keep the participant
on the study intervention under an alternate mechanism as necessary.
o If a participant is incarcerated temporarily while enrolled in a study, and if that incarceration
has no effect on the study, the participant shall remain enrolled.

Table of Contents
Page 235 of 273
o If the temporary incarceration has an effect on the study, the IRB shall handle the case
according to the case set forth above.
o Note: An adolescent (e.g., age 14) detained in a juvenile detention facility is a prisoner
therefore; the IRB would need to comply with Subpart C and Subpart D – Children.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
10/14/2021
Replaced Julie’s name with Carol in the payment section and archived log of updates over 1 year old

Table of Contents
Page 236 of 273
SOP Title: VA Studies with non-VA Sites – IRB Submission Requirements
SOP Category:
Study Management
Established:
4/11/2019
Last Revision:
4/11/2019
PURPOSE
The purpose of this document is to describe the additional IRB submission requirements for study where
the sites include both the Atlanta VA and Emory University or its affiliates, without a PI credentialed at
both sites.
SCOPE
The SOP applies to studies with both a site at the Atlanta VA and a site at Emory University or its
affiliates, without a PI credentialed at both sites.
RESPONSIBILITIES
• VA liaison: IRB analyst in charge of guiding teams through the process of submitting a VA study via
the eIRB system
• Study team: in charge of making revisions to their submissions and to follow the VA liaison
instructions
• IRB Director: receives notification from VA liaison and assigns non-VA version of study to RPA with
additional instructions
• Emory RPA: collaborates with the VA liaison to make sure that both studies are reviewed at the
same meeting, if possible
PROCEDURE
• If consulted before approval, the VA liaison will let study teams know that the PI of a study taking
place at the Atlanta VA must be credentialed at the VA. If the overall study PI is not so credentialed
at VA, there must be a separate VA credentialed PI (local site investigator) for the VA site. In general
a separate eIRB submission is preferred for that PI/site; however, if the VA credentialed PI is listed
on the grant application and the data is to be combined as part of a collaborative project, an
exception can be requested through the VA Research Office and the IRB Director.
• If the VA liaison is assigned a study that, despite the above, takes place at both VA and non-VA sites,
and indicates that there two separate PI’s, (for example, via listing different PI names in the consent
forms) or has a single PI who is not credentialed to serve at the Atlanta VA, he will log the following
as a comment in the study history:
o “Dear study team: My name is X and I will be conducting the initial review of your
study. I noticed that this study has [non-VA site(s)] and the Atlanta VA as sites, but does
not have a PI credentialed to conduct research at both sites. If this is correct, we
require that you submit separate submissions for each site, as the Atlanta VA requires a
PI with valid VA credentials. Please confirm if this is your case, and provide the name of
the person serving as PI at the VA, and a research coordinator. To prevent additional
work on your part, we will clone the study and provide it to you in “pre-submission”
status so it can be submitted to the correct department head right away. Make sure
you specify in the protocol the work that will be done at each site, so it is clear for both
submissions. In the event that this is a collaborative study and you wish to continue
with one IRB submission, you will need to request a consult with the VA Human

Table of Contents
Page 237 of 273
Research Protection Group. The Group will discuss with the IRB you request and make a
determination if the systems are in place to properly manage this as one IRB submission.
Let us know if you have any questions”.
• In the event that the study requires two submissions, and after obtaining the names for the PI and
coordinator for the VA site, the VA liaison will do the following:
o Click on “clone study”. The system will ask for a name and they should name the study
“Emory-CURRENT_STUDY_NAME”.
o Locate the newly created study. You can locate the newly created study by searching
for the short title under the Projects tab using the name assigned to the new study.
o Once you navigate to the newly created study, click on “Admin View”
o Put the study back in “Pre-submission” under “Status”, and change the name of the
study PI and coordinator.
o Log a comment in the new study indicating that this study should be reviewed
concurrently with the VA version of the study (provide IRB study number)
o Once the study is submitted to the IRB, the IRB Director will let the new study’s RPA
know that this study is similar to one submitted for the VA, and instruct the RPA to
communicate with the VA Liaison.
• The RPA will let the VA liaison know the Emory study has been submitted, and to ask him which
meeting will review the VA study, if already known, and attempt to assign it to the same meeting. If
not already assigned, RPA and VA Liaison should work together to have the studies reviewed at the
same future meeting., with Agenda Notes indicating that they are the same protocol at two
different sites
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES

Table of Contents
Page 238 of 273
SOP Title:
Determinations and Reviews by IRB Staff
SOP Category:
Study Management
Established:
6/26/2012
Last Revision:
2/1/2019
PURPOSE
To detail the type of determinations and reviews that can be made by a qualified IRB staff. The IRB
Director shall decide when IRB staff is qualified, based on length of experience and demonstrated
expertise.
SCOPE
New studies, continuing reviews, and Modifications submitted to the IRB office for review.
PROCEDURES
Reviews Permitted by IRB Staff (without involving an IRB member):
1. Determining whether a study qualifies as research involving human subjects (NR/NHSR)
2. Determining whether a study should be reviewed as Exempt
3. Approving Exempt reviews that do not involve HIPAA Privacy Rule waivers or authorizations. Only
staff authorized by the IRB Director to approve exempt studies can do so. For more information
about exempt research that can be reviewed by staff or staff designated reviewers, review
this
guidance.
a. Modifications: similar to new exempt study submissions, Modifications submitted for
exempt studies can be reviewed only by staff authorized by the IRB Director to approve
exempt studies.
4. Approving certain minor administrative changes.
5. Approving an Modification submitted only to transition a study to the Revised Common Rule-no
other changes requested. If the Modification is also updating the consent form to align with the new
elements in the Revised Common Rule, that Modification should be reviewed by an IRB member
(staff or faculty member).
Minor Administrative Changes: Some minor administrative changes may be approved by qualified IRB
staff. These changes are exclusively limited to the following:
• Change of contact information
• Addition or deletion of junior level personnel (not co-investigator, principal investigators)
• Addition of co-investigator
• Addition or removal of co-investigator for minimal risk studies
• Title change that does not reflect a change in study
• Corrections of typographical errors
• Reformatting of unchanged text
• Errors in completion of the IRB application, as confirmed with study staff as appropriate (as long as
the study was initially reviewed with the correct impression)
• Removal of study sites that were never activated
• Change of funding status from “pending” to “approved” (if federal grant application was previously
approved as pending by the IRB, and it is not a new funding source)

Table of Contents
Page 239 of 273
REFERENCES
• Emory IRB P&Ps, chapter 28 (IRB Protocol Triage and Assignment of Review Category)
• Emory IRB P&Ps, chapter 30 (Exempt Research)
• Emory IRB P&Ps, chapter 40 [Protocol Modifications (Modifications)}
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/14/2015
Added a Purpose and Scope sections.
9/16/2016
Added P&P reference
5/5/2017
Clarified the type of changes in funding staff can approve
1/20/2019
Adding that staff can review Mod only requesting the transition of a study to the Revised Common
Rule if it does not include any other changes but the transition.
2/1/2019
Added that Mod to exempt studies can be reviewed by staff authorized by the IRB Director

Table of Contents
Page 240 of 273
SOP Title:
Categories of Research Reviewable by IRB Staff as IRB Designated Members
SOP Category:
Study Management
Established:
3/29/2013
Last Revision:
4/23/2024
Background/Policy: IRB Staff can perform reviews within the scope described below only if they are
appointed as IRB members, and have been designated by an IRB Chair as authorized to perform
expedited reviews.
Note that all IRB staff, including those appointed as IRB members, can review minor administrative
changes as described in the IRB Policies and Procedures, “Modifications” chapter, and the SOP entitled
“Determinations and Reviews by IRB Staff”.
A. NEW STUDIES
1. Minimal risk expeditable studies involving only the following:
a. F (5) (e.g chart reviews or specimen analyses, whether retrospective or prospective)
b. F (7), but only as a last resort when faculty DRs are particularly busy or unavailable.
Before such an assignment can occur, the study analyst should consult a TL or Director
to make sure there are no other available reviewers to help. In addition, the staff DR
should have enough expertise to be comfortable with such reviews.
If staff IRB member is not comfortable with children populations or sensitive studies, forward to
faculty DR.
2. Exempt Studies: Staff DRs can review all exempt categories. If a DR is uncomfortable reviewing
any category of research, the staff DR can reassign to faculty.
a. Remember: Do not use D4 for chart reviews because our HIPAA policy does not cover
these type of studies. Use F (5) instead. Also, For more information about exempt
research that can be reviewed by staff non-DRs or staff designated reviewers, review
the ‘Guidance – Non-Committee Reviewers’ document in H:\General\Admin IRB
Documents\Checklists-Staff, Forms, and Templates\1. Staff Screening Checklists and Tip
Sheets.
3. Contingency reviews (full board and expedited studies):
a. ICF revisions as specifically requested by IRB Committee
b. Smartform revisions as specifically requested by IRB Committee
c. Confirmation of full board’s stated assumption (i.e. not new information from study
staff)
Note that all IRB staff can do contingency reviews for Cost and In Case of Injury options, as well as
ancillary committee approvals where no changes were required.
B. MODIFICATIONS
1. Changes of any kind to studies that Staff DR could have approved at initial review (see the
section above), unless the changes move the study outside that scope. For example, F5 studies,
moving to other review categories due to the addition of procedures.
2. Changes to informed consent that are made solely to update template language
3. Changes to PI on minimal risk studies where the PI is NOT the IND/IDE holder
4. Deletion of Co-Investigators on more-than-minimal risk studies

Table of Contents
Page 241 of 273
a. Deletion of Co-investigators: inquire about the replacement of expertise
5. Updates of other documents insofar as they are needed to reflect changes 2-4 above
6. Change to ICF, protocol, or other study documents that are limited to corrections or factual
updates (i.e. no changes to protocol risk, benefit, design; not clarifications to parts of the
protocol that were not clear); simplification for lay-friendlier language, or reflecting completion
of a pending issue the IRB knew was in process (such as Cert of Confidentiality, funding). (See
“Minor Administrative Changes” list in Emory IRB P&Ps for a subset of
corrections/factual/administrative updates that all IRB staff may review.)
7. Use of electronic informed consent when:
a. Obtaining an electronic signature using an Emory LITS approved method, if the consent
content was already approved by a faculty designated reviewer or the IRB Full board.
b. Obtaining verbal consent, when the verbal consent content and verbal consent process
has been already approved by a faculty designated reviewer
8. Adding funding sources (but not delete them)
9. Adding Emory-affiliated sites (includes EHC, CHOA if not also adding minors for the first time, St
Joseph’s/Johns Creek, Grady)
10. Expeditable increase or decrease of N if the same type of population already approved, OR to
allow more consents/screen failures - both on MIN RISK studies only (P&Ps say what levels of
increase are expeditable). If new population or more than min risk, send to faculty.
11. Changes in data collection instruments (questionnaires, surveys, chart abstraction forms, CRFs)
as long as expertise is not needed to assess risk-level change
12. Advertisements and other recruiting materials
13. Retention materials, newsletters, blasts, reminder cards, health tips related to the disease being
studied, participant alert cards, other miscellaneous items that do not involve changes to
compensation,
14. Sensitive study determination requests, if not done when the application was initially submitted
15. Translated versions of approved documents, requests to enroll non-English speaking subjects,
and the addition of Short Form consents in other languages if the population to be studied (for
example, patients with leukemia) are the same.
C. CONTINUING REVIEWS
1. All chart reviews, secondary data analyses, and research on existing specimens - F(5)
2. All F(7)
3. Data analysis only (DAO):
a. For full board studies (i.e. F8c) – but not the first year that study has entered DAO
stage. Those should go to faculty reviewer because there may have been significant new
information in the past year before subject interaction ended.
b. For all other categories of expedited studies with no restriction on how long the study
has been in DAO.
D. REPORTABLE NEW INFORMATION SUBMISSIONS
1. Acknowledgments per Associate or Assistant Director review (based on Emory IRB P&Ps)
2. Finalizing decisions made by Compliance Review team or chairs (document)
STAFF DRS REVIEWING THEIR OWN ITEMS:
• CAN review their own:

Table of Contents
Page 242 of 273
o Continuing Reviews and Modifications that would fall under “minor administrative changes”
per P&Ps
o Truly retrospective F(5) new studies
• CANNOT review their own:
o Exempt studies (need to obtain a second opinion on exempt status)
o Prospective F(5) new studies (need to get a second opinion on consent/HIPAA waivers)
In general, all staff should consider the benefit of sending other Modifications to another reviewer but
may use their own judgment.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/11/2015
Clarified when staff DRs should send their own submissions to others instead of reviewing for
themselves (end of SOP), clarified what kinds of “data analysis only” Staff DRs can handle (now
includes all expeditable studies as well, not just F8c).
2/18/2015
Clarified what Data Analysis Only renewal reviews can be done by staff DR
4/2/2015
Added that Staff DR can approve new Emory-affil study sites. Also CR’s F(5) applies to secondary data
analysis in general, not just retro chart reviews. Also reworded Mod’s for increase N to make clearer.
2/18/2015
Changed title (Designated REVIEWERS, instead of Designated MEMBERS) and added
Background/Policy. Clarified Modifications section. Removed any listing of items that all IRB staff can
actually review. Can now review adding Grady as site. Took off items that actually all IRB staff can
approve (minor admin changes).
9/16/2016
Minor corrections
12/20/2016
Clarified that Staff DR’s can review all expedited studies in Data Analysis Only (had been stated
already, but not clearly)
1/20/2019
Added information for Exempt Categories review-Revised Common Rule
2/1/2019
Provided more clarification about exempt category review, specifically studies that should be sent to
a faculty reviewer instead than to a staff member.
6/14/2020
Adding that staff DRs can approve the use of eConsent in studies, as long as the consent content or
consent process (in the case of verbal consent) has been previously approved by the FB or faculty DR
4/23/2024
Clarified details regarding ability to review studies deemed sensitive among other minor updates

Table of Contents
Page 243 of 273
SOP Title:
Assignment of New Studies to IRB Staff
SOP Category:
Study Management
Established:
2/9/2018
Last Revision:
8
/15/2024
PURPOSE
The purpose of this document is to detail the steps involved when newly submitted studies are assigned
to an IRB analyst.
SCOPE
The SOP applies to studies submitted to the Emory IRB.
ROLES
Analyst – receives assigned studies.
IRB RPA Team (currently the IRB Director, IRB Associate Director, IRB Assistant Directors, and IRB Team
Leads) – receives daily assignment email; able to perform spreadsheet maintenance.
IT RPA Specialist (currently Kiran Isukapatla) – Bot creator; available for consultation when performing
maintenance.
PROCEDURE
A Robotic Process Automation (referred to as “Bot”) was created to assign incoming new study
submissions in eIRB to IRB Analysts. This runs daily and there are pre-determined rules that dictate who
receives each assignment. The Bot runs a custom report in eIRB (ORAIT_IRB New Study Assignments_v2
)
to generate a list of studies that have been submitted to the IRB over last 24 hours and have not yet
been assigned to an analyst.
1. Current IRB analysts are placed into one or more teams on the “Analyst Breakdown Sheet
” and will
receive new study assignments automatically per the following rules (see sections further below for
information about maintaining the spreadsheet):
• Reliance Team
o If question 5 in “Basic Study Information” is answered “Yes” (question: Will an external
IRB act as the IRB of record for this study?); or
o If question 6 in “Basic Study Information” is answered “Yes” (question: Will your IRB act
as the single IRB of record for other participating sites?)
• Q Team
o If question 3 in the “For Clinical Research/Expanded Access Only” section is answered
“Yes” (question: Is this an expanded access submission for an unapproved drug or
device?)
o If question 1 under device is answered “Yes” in response to “Is this a humanitarian use
device (HUD)?”.
• Sponsor-Investigator Team
o If “investigator” is selected as the IND Holder (for drugs) and/or the HDE/IDE Holder (for
devices).

Table of Contents
Page 244 of 273
• AVAMC Team
o If Atlanta VA Medical Center is listed as a Local Research Location.
• Chart Review Team
o If “Complete Waiver of HIPAA Authorization (because it is impracticable to obtain signed
HIPAA authorization from participants)” is selected under question 3 in the “HIPAA
Applicability and Waivers Requested” section; and
o If “No informed consent will be sought (e.g. in retrospective chart review with waiver)”
is selected under question 1a in the “Informed Consent Process and Waivers
Requested” section; and
o If “Waiver of any or all elements of the informed consent or assent process/form” is
selected under question 2 in the “Informed Consent Process and Waivers Requested”
section; and
o If “Is this an "applicable clinical trial" or a study that otherwise requires registration in
ClinicalTrials.gov?” in the “For Clinical Research/Expanded Access Only” section is
answered “No” or left blank.
• Regular Team
o Any other study requiring Emory IRB review and approval.
2. After a study is assigned, the Bot will add the below comment directed towards the study team
selecting PI/PI Proxy/Primary Contact under question 3, “Who should receive an email
notification?”:
“Thank you for your submission! It has been assigned to the study analyst listed above under “IRB
coordinator”. They will perform a pre-review screening of the study and let you know if any changes
are needed before this study is sent for further review.
Our target turnaround time information is here:
https://irb.emory.edu/about/services/irb-target.html
Please reach out to your analyst with any study-specific questions.
Please note: If you have not used our required protocol templates available on our website (see link
below), this will be required so you may wish to start working on that while your analyst does their
pre-review screening. You can upload the revised protocol after the analyst sends the submission
back to you.
Note: For multisite studies not led by Emory, you should submit the multisite protocol but also an
Emory supplement using our “Supplement” protocol template.
https://irb.emory.edu/forms/protocol-templates.html
”
3. The Bot updates the “All Analysts” tab in the “Analyst Breakdown Sheet
”
4. The Bot sends an email to the IRB RPA Team listing that day’s study assignments
a. The recipients review the daily spreadsheet to ensure the Bot is working correctly

Table of Contents
Page 245 of 273
5. If any studies are manually assigned or reassigned by IRB staff: request IRB RPA Team help with the
procedure in “Spreadsheet Maintenance” section below, under “As Needed.”
Structure and Function of Assignment Spreadsheet:
The IRB Analysts are listed on the relevant team tabs in the “Analyst Breakdown Sheet
.” Assignments
are made on a rotating basis considering each Analyst’s prior assignments for that week, their current
availability, and their assigned team(s). A weekly total is tallied based on all assignments to a specific
Analyst and not broken down by each group they are part of(tallied under the “All Analysts" tab).
Steps to temporarily or permanently change Analyst groupings or availability status are in the sections
below.
Spreadsheet Maintenance Procedures – IRB RPA Team Only:
Weekly (Fridays)
• Every Friday after the Bot has assigned studies, the “Analyst Breakdown Sheet
” will be updated
by a designated member of the IRB RPA Team. The information in the “All Analysts” tab should
be copied and pasted into an archive tab created for the current week (e.g. “Week of XX-XX-XX”)
for a historical record. The total assignment number should be tallied below the assignments for
reference.
• Next, the assignment loads for the week should be cleared out for the upcoming week, on the
“All Analysts” tab. In addition, any Status column notes for the coming week (staff that will be
OOTO or shouldn’t receive assignments for a period) should be added or removed at this time.
As Needed
• Availability updates: If there is a reason an Analyst should not be assigned studies for a given
period (e.g. vacation, sickness, meeting facilitation week), a note must be added to the “Status”
column in the “All Analysts” tab of the spreadsheet
. When any character is included in the
“Status” column for an Analyst, it will prevent the Analyst from receiving assignments.
o The specific language in the comment has no effect on the Bot; it is not smart enough to
know how many days the Analyst will be unavailable. The text is just for IRB RPA Team
reference.
o Delete all text in the cell once the Analyst should receive assignments again.
• Changing Analyst Teams/Roles: Analysts can be moved to different teams if roles change.
• Adding or removing IRB Analysts (for new hires/departing staff): To add a new employee or
remove a past employee, the edits need to be made to the specific team tab and the “All
Analysts” tab, including the “Member” column that lists the full analyst’s name (if this column is
not updated, assignments will not be tallied correctly).

Table of Contents
Page 246 of 273
• Important: When a study is manually re-assigned after the Bot assigns it, the total assignment
count must be manually updated for both Analysts (the one originally assigned and the one to
whom it was reassigned), and a note left in the “Notes” column of the ”All Analysts” sheet. The
note should include the Study ID for the submission that was reassigned. When a study is
manually assigned before the Bot runs, the total assignment count must be manually updated
solely for the assigned Analyst, and a note left in the “Notes” column, with the study ID.
Things to consider:
• The rules aren’t perfect and are not sensitive to erroneous or inconsistent information provided
in a submission.
• New study assignments should be reviewed by the assigned analyst in a timely manner to
determine if they require re-assignment based on the information in the submission. Some
important information to consider:
o Is this an industry-sponsored, Ph I-IV industry-sponsored, industry-designed drug or
device trial, it should be reassigned to the Reliance Team and the total count updated,
as it requires review by a commercial IRB.
o If a study, other than a chart review, is assigned to the Chart Review team it should be
reassigned to an analyst on the regular analyst team and total count updated.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/31/2019
Replaced “reliance specialist” with “reliance RPA”
01/11/2023
Extensive updates for evolving process
8/15/2024
Overhauled for the new Robotic Process Automation implemented to assign incoming
submissions to the IRB.

Table of Contents
Page 247 of 273
DURING STUDY CONDUCT
SOP Title:
Over-Enrollment Via Consent (No Research Activities including during Screening)
SOP Category:
Study Management
Established:
5/15/2012
Last Revision:
6/9/2021
PURPOSE
The purpose of this SOP is to explain the process to follow if over-enrollment took place in a study, when
the subjects enrolled beyond informed consent (hereafter referred to as “over-enrollment via consent”).
It may also be used if researchers also did basic screening with those subjects, i.e. reviewed those
subjects’ charts or asked them for private information.
SCOPE
This SOP only applies when over-enrollment was due to consenting more subjects than had been
approved by the IRB. It does not apply to cases in which the over-enrollment included subjects who
completed any study-driven interventions (even if done for screening or if the activities are considered
minimal risk), or post-screening study activities. These activities may include fasting for blood test and
medication withdrawal before informed consent was obtained.
RESPONSIBILITIES:
• IRB Analyst: log over-enrollment in log located under the QA folder (see folder name below)
• IRB Team Q designated member: will periodically trend the data in the spreadsheet to identify
potential continuing noncompliance and send monthly reports to CoRe.
PROCEDURE:
Cases should be maintained in a running record sorted by PI. No need for CoRe to review individually
unless there is evidence of continuing noncompliance. Team Q is responsible for reviewing records to
identify trends of persistent over-enrollment. Persistence of over-enrollment after the issue has already
been identified and pointed out to the PI could be continuing noncompliance and should be sent to
CoRe. Team Q will review the spreadsheet monthly and report all instances of potential continuing
noncompliance to the CoRe team. Other general trends (e.g., departmental patterns) will be reported to
the CoRe team and the Associate or Assistant Directors.
Analyst
If enrollment is close to the cap, but not over it, analyst should recommend a review and, perhaps, an
increase in enrollment cap. If an analyst identified a case (e.g. at continuing review), he/she will contact
the PI providing:
• A description of the noncompliance (the number approved vs. the number consented
• A corrective and preventive action plan that includes:
o The submission of a Modification to increase enrollment (if the study is closed to
enrollment, the NC should be acknowledged, but no Modification is necessary).
o Portfolio-wide audit of enrollment numbers (See Guidance to PIs Regarding Self-Audits)

Table of Contents
Page 248 of 273
o Education by the analyst about the issue with references to IRB P&Ps (p. 155 defines
enrollment in Chapter 43, Informed Consent, under Procedures)
The analyst should follow up to make sure the Modification is submitted (using Outlook calendar or
other ticklers). If it is not submitted by the time of review, it becomes a pending issue. The analyst
should enter the issue into the over-enrollment tracking sheet. The tracking sheet is in the QA working
files main folder. H:\IRB\General\QA Working Files
Guidance to PI’s Regarding Self-Audits
Paste as a Logged Comment:
• If over-enrollment via consent is not present in any other studies, there is no need to report this.
• If over-enrollment via consent is discovered in other active studies, please submit a Modification
to increase enrollment and explain why (i.e. discovered as part of audit).
• If over-enrollment is found that was not due to solely to consenting more subjects than had
been approved but included exposing subjects to screening or procedures beyond the consent,
report the over-enrollment as a protocol deviation in the Reportable new information
submissions module for the specific study in eIRB.
-On the “Protocol Violation/Deviation” page, check yes for question #1, “Is this a
substantive deviation from the protocol as approved by the IRB?”
-For the second question, indicate that the deviation/violation adversely affects: the
rights/welfare of subjects and the safety of subjects
PROCESS FLOW
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
9/16/2016
Added clarification about screening procedures
10/26/2017
Clarified purpose of SOP (including title)
6/9/2021
Added additional info to clarify that only active studies are required to be audited for over-
enrollment
Analyst identifies issue
and adds to log
Analyst communicates
with PI and logs
comment per
instructions
Team Q reviews log
Team Q sends info to
CoRe if applicable

Table of Contents
Page 249 of 273
SOP Title:
Transferring Study Participants Between Study Sites
SOP Category:
Study Management
Established:
11/10/2010
Last Revision:
10/26/2017
PURPOSE
To explain the steps to be taken if a study is transferring data or patients between sites
SCOPE
Studies approved by the Emory IRB
ABBREVIATIONS
• OTT: Office of Technology Transfer
• OEC: Office of Ethics and Compliance
• DTA: Data transfer agreement
DEFINITIONS
• Data transfer agreement: agreement used when transferring protected health information (PHI),
including limited data sets, from one party to another.
PROCEDURE
For Patient transfer
• Study team should refer to the OTT/OEC guidance for data transfer agreements. The information can
be accessed at http://www.orc.emory.edu/hipaa/index.html
.
o Outgoing DTA: To initiate routing and initiation of the appropriate DTA, Emory investigators
should complete an Outgoing DTA Questionnaire (provided at
http://www.med.emory.edu/research/resources/index.html
or
http://www.ott.emory.edu/forms/index.html) and email the questionnaire to
somdta@emory.edu.
o Incoming DTA: OTT manages all DTAs covering receipt by an Emory investigator of human
subject data to be used for research purposes not involving a clinical trial or human subject
interaction/intervention by the Emory investigator. All OTT-managed incoming DTAs should be
forwarded to OTT for approval, along with a completed Incoming DTA Questionnaire (see
“Incoming DTA Questionnaire” on the OTT website at
http://www.ott.emory.edu/forms/index.html
). Faculty members should forward these
documents via email to m[email protected].
o Note: data must not be transferred until the subjects who are transferring have signed the new
HIPAA authorization
o Any subjects currently on study have to be notified about these changes and offered the option
of transfer or study termination, with appropriate referrals if needed.
• Study team should submit a Modification to the Emory IRB for a revised or new HIPAA authorization
to cover the transfer of PHI from Emory to the new site for research purposes.
The requirement to

Table of Contents
Page 250 of 273
complete an appropriate DTA for a transfer of human subject data is in addition to, and not a
substitute for, obtaining appropriate applicable informed consent, HIPAA authorization, or IRB
approval for any research activity. Consult with AVP or a Director for information about the HIPAA
form information.
o Subjects will need to sign both a new ICF and a new HIPAA authorization with the other site.
o Study team should close out the study with the Emory IRB after all the transfer activity has been
done and no more activity with PHI is active.
*This SOP will also apply if subjects transfer to a new site without a study closing at Emory (e.g. because
the subjects are moving far away and there is another site closer to their new home)
For Data Transfer
• Study team should refer to the OTT/OC guidance for data transfer agreements. The information can
be accessed at http://www.compliance.emory.edu/hipaa/index.html
.
o Outgoing DTA: To initiate routing and initiation of the appropriate DTA, Emory investigators
should complete an Outgoing DTA Questionnaire (provided at
http://www.med.emory.edu/research/resources/index.html
or
http://www.ott.emory.edu/forms/index.html) and email the questionnaire to
somdta@emory.edu.
o Incoming DTA: OTT manages all DTAs covering receipt by an Emory investigator of human
subject data to be used for research purposes not involving a clinical trial or human subject
interaction/intervention by the Emory investigator. All OTT-managed incoming DTAs should be
forwarded to OTT for approval, along with a completed Incoming DTA Questionnaire (see
“Incoming DTA Questionnaire” on the OTT website at
http://www.ott.emory.edu/forms/index.html
). Faculty members should forward these
documents via email to m[email protected].
• Note: the IRB staff should let study team know but the submission process (Modification, new study)
should continue.
• The approval letter should indicate that data must not be transferred until the agreement is
completed.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
10/3/2014
Updated information after OTT/ORC revised guidance
2/15/2017
Changed title and move comment to the end to avoid confusion about subject matter covered by
SOP
10/26/2017
Adding information about data transfer, purpose, definitions, etc.

Table of Contents
Page 251 of 273
SOP Title:
Fixing Errors in eIRB System
SOP Category:
Study Management
Established:
5/8/2020
Last Revision:
11/3/2020
PURPOSE
To explain how to fix errors in the system after a study submission is approved
SCOPE
Studies approved by the Emory IRB
PROCEDURE
Studies approved in the new eIRB system
• As the study owner (or if you have the “Director” role in eIRB) you can make corrections to the
record. Make the changes by clicking on “submit designated review” or “submit committee review”,
according to the study approval route (DR vs. FB). Taking this action will allow for fixing errors
involving approval/expiration dates or when the correct documents were not finalized.

Table of Contents
Page 252 of 273
After the “Submit Committee Review” or “Submit Designated Review” options are executed, other
errors can be fixed by clicking on “Edit Pre-Review”.
Studies migrated from RX (old eIRB)
IF THE STUDY SHOWS AS “LAPSED”, THE STUDY TEAM WILL NEED TO SUBMIT A CONTINUING REVIEW SUBMISSION INSTEAD
OF A MODIFICATION.
• The preference is to ask the study team to submit a modification submission but, as stated above, a
CR may be required to change an incorrect study status.
• If submitting a modification:
o The study team should select “other parts of the submission”, and should not include any
additional changes to the mod.
o If a study team has a modification already open, we will have to wait until that modification is
completed to fix the error if the error would involve re-stamping documents and/or fixing
approval dates.
o If the error would not involve re-stamping documents/fixing dates, then the same modification
can be used. If so, make sure the approval letter documents the additional changes made in the
submission to fix the error.
• If submitting a continuing review:

Table of Contents
Page 253 of 273
o The study team should not worry to include new data in the submission as this is only done for
admin purposes.
• Let the team know that this submission is only going to fix the error, and will not be an approval of
additional documents, as specified above.
• For modifications or continuing reviews submitted anew to fix errors
o After the team has submitted the application, you can approve it using the “designated review”
route.
o Select “expedited review” and under the category of review, select “other”.
o Under question 7 (notes), explain that this submission is not approving any change/renewing
the study but it is submitted only to be able to fix an error and specified what is being fixed.
o When working on the approval letter, select the “Approval of Modification” letter. The title of
the approval letter for this submission should read “Administrative Submission to Correct ___”.
In the body of the document, if applicable, we should state that the document being stamped
was approved on DATE. The document will be stamped with today’s date as the system does
not allow for retroactive stamping.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
5/18/2020
Adding that a CR is needed when the study is showing as “Lapsed”.
11/3/2020
Making additional changes to the SOP to reflect the current thought process

Table of Contents
Page 254 of 273
SOP Title:
Continuing Review Processing-Preliminary Analysis through Approval
SOP Category:
Study Management
Established:
6/28/2011
Last Revision:
5/5/2021
PURPOSE
The purpose of this SOP is to describe the process that research protocol analysts (RPAs) will follow
when processing continuing review (CR) submissions through approval. It includes the process for
transitioning studies to the revised common rule.
SCOPE
The SOP applies to CRs for single-site studies. (See Collaborative Research SOPs for processing multi-site
studies where Emory is the single IRB of record.)
Menu
• Studies wishing to transition to the revised common rule
• CR Screen Process Pre-review
• Expedited Review
• Full Board Committee Review
• Finalizing Submission
PROCEDURE
1. Use this SOP and open the Continuing Review Worksheet. Use these documents together when
reviewing CRs.
2. For technical issues, refer to the Huron Staff Quick Reference
tool.
CR Screen Process:
1. Open the CR review worksheet located in in this folder
, H:\General\Admin IRB
Documents\Checklists-Staff, Forms, and Templates\1. Staff Screening Checklists and Tip Sheets
2. To see all CRs assigned to you in your inbox, including CRs you have sent back to the study team for
clarification, run your CR Report following the steps below. Use the Huron IRB Staff Quick Reference
for technical information.
a. In eIRB click on “Reports” on the tab at the top of your page.
b. Click “Custom Reports”

Table of Contents
Page 255 of 273
3. At the bottom of the page change 10/page to 100/page so you can see all options.
4. Page down to ORAIT_CRs and select this option.
5. When the Search Results window opens, click Pop Out at the bottom of the screen.
6. In the Filter By drop down menu, select “IRB Owner”.
7. In the filter box enter your last name.
8. Click the magnifying glass icon.
9. At the bottom of the page change 10/page to 100/page to ensure you see all of your items.
10. You can click Study Date Expiration or Project State to sort items.
11. Review the Huron Staff Quick Reference
for eIRB technical steps starting on sections “Complete a
Pre-Review”.
12. If clarifications are needed or items are needed from the study team, click on “request pre-review
clarification” in the left menu. You can find technical steps on page 15 of the Huron Staff Quick
Reference. Use page 16 to compare the documents you are receiving, if any. If the CR will be
expedited, all clarifications must be resolved including CITI training and modifications to remove
study staff approved before assigning to the reviewer.
13. Follow up with the study team if a response is not received in a timely fashion. If a submission is in
requested changes for more than 30 days with no answer from the study team, the analyst should
withdraw it. The analyst needs to document their efforts in contacting the study team and email
them before withdrawing the study.
14. If the CR will be reviewed by the full board, the CR must be in pre-review (not clarifications
requested) before submitting the pre-review. The CR can be assigned to a committee, and CITI
certificates and DSMB reports can be uploaded with a comment. If there is a modification to remove
study staff, approve it before finalizing the CR.
a. Note: If requesting a current DSMB report, give the study team a deadline to provide it so
the committee members have a chance to review it.
15. To process a modification to remove study team members, follow the SOP titled “Adding/Removing
Study Personnel.”
Pre-review:
1. Click “Submit Pre-review”. For technical instructions, review the Huron Staff Quick Reference
, on
page 14. A new window will open.
a. Question 1 - For studies under the pre-2018 common rule, the first question will be the
Common Rule Requirements (see below). There is no mechanism to change this answer, so
once a study has transitioned to the revised common rule, it cannot be moved back.
Confirm the study is not FDA regulated study or under DOJ funding. For more information,
see the section of this SOP below titled “Studies wishing to transition to the revised
common rule.”
b. For questions 1-4 (or 2-5 if your first question was about transitioning to the revised
common rule),

Table of Contents
Page 256 of 273
• confirm the boxes that are checked are correct based on the information you have
reviewed about the study. Refer to the SOP titled “Pre-Review Options and Ancillary
Review Information.”
1. If the information is already populated, do not make any changes to the Pre-
Review options without confirming with a Director or AD unless this
submission is a modification/CR combined.
2. If there are no boxes checked, check the eIRB record for applicable selection
• Add missing materials and notes, if any.
2. Click Yes to the last question to complete the pre-review. (If you know you will need to edit the pre-
review later, mark No.) Pre-review must be complete before routing to a designated reviewer or
assigning to a committee.
3. For information about the required selections under Pre-review or verifying appropriate ancillary
reviewers, please review this SOP
If the selections are already made, just leave the selections from
the previous review (unless this is a MOD/CR with changes to the study).
4. If the study shows as transitioned to the 2018 rule but this is not correct, log a comment to the
reviewer explaining this discrepancy, so they do not say the study does not require a CR in the
future.
Transitioning studies to the revised common rule:
Check this information
to learn what we mean by the pre-revised common rule and the revised common
rule.
Studies required to transition per the guidance found in the link above (under “Emory IRB Transition
Plan”) should be transitioned using the CR submission.
1. Review the answers to the CR, ensuring the study is not FDA regulated or DOJ supported.
2. Follow the instructions below under “Pre-review” section 1a.
o Click “Submit Pre-review”
o Select 2018 requirement, so the study can transition after the CR is approved.
o If it is not clear if a study is FDA regulated, consult with the Director. Remember than any drug,
device, biologic, software or app (considered devices) are FDA regulated even if previously FDA-
approved.
3. For studies wishing to transition: The study team needs to submit the
Guidance and Attestation
form with the continuing review. If the study is still enrolling subjects, they need to revise the
consent to align with the revised common rule requirements by submitting a modification (MOD).
o If the study requires a MOD (as they are still enrolling subjects), and the MOD has not been
submitted yet, ask the team to withdraw the CR and resubmit a MOD/CR application when the
changes to the consent are ready. The only way to transition to the revised common rule is via a
CR submission, so if the study is close to expiration, we may need to work on the CR submission
and ask them to submit a MOD/CR after approval if still wishing to transition.
o The study owner should be the person in charge of screening the MOD/CR application.

Table of Contents
Page 257 of 273
Expedited Review
1. Select the reviewer based on their specialty, if possible. For example, give cancer, sickle cell, or
infectious disease studies to the reviewers who specialize in those areas. If choosing a Staff
designated reviewer, make sure you follow the SOP entitled: “Categories of Research Reviewable by
IRB Staff as IRB Designated Members”. Follow the Huron Staff Quick Reference
, page 22 for
additional steps.
2. Send the CR to the reviewer at least 3 weeks before expiration (if possible due to CR submission).
When assigning, you may suggest review categories based on your review and the prior years’
reviews. Make sure that you do not imply that is the category to be used as the member should do
their own review. Add this information under question 2 when assigning to a reviewer. If the
category from previous years is obviously wrong or not listed, contact your TL or Director.
3. After the CR has been reviewed, follow the post-review activities detailed on Page 25 of the
Huron
Staff Quick Reference. If the CR is approved (no clarifications are needed) skip to the section of this
SOP titled “Finalizing Submission.”
4. If clarifications are requested by the designated reviewer, draft a letter to the study team with the
required changes.
5. Follow up with the study team if a response is not received in a timely fashion. If a submission is in
requested changes for more than 30 days with no answer from the study team, the analyst should
withdraw it. The analyst needs to document their efforts in contacting the study team and email
them before withdrawing the study.
6. Once a response is received, click on “submit designated review” and send to the original reviewer.
Let them know this is an additional review to confirm that the previous changes they requested
were completed.
7. The state will change to “Post-Review.” Review the dates to confirm they are correct. If not, correct
them before proceeding by clicking “Submit Designated Review.”
8. You are now able to generate a letter for the approval of the CR application.
Full Committee Review
1. If the CR requires review at the full board, assign it to the appropriate committee agenda four weeks
before expiration (if possible).
a. Click “Assign to Meeting” and choose from the options. Choose a committee date that is ideally
a couple of weeks before expiration (one week may not be enough, as there may be pending
issues) and choose a committee that has appropriate expertise if needed.
b. Click on “Edit Pre-review” and add the following information under “Notes”:
a. Whether enrollment numbers are compliant with IRB approved enrollment numbers in the
protocol.
b. If there are any CITI/training pending issues that may prevent study reapproval
c. If this is a Sponsor-Investigator study:
a. Indicate whether the S-I responsibility form is included in the submission with an S-I
consult approval (This is required before the CR can be approved) (if not, pending
issue)
d. If there are any Committee member conflicts you are aware of
e. If it is not clear why the CR is going to full board, explain (e.g. list research interventions still
ongoing)
NOTE: You are required to update the pre-review no later than the Monday before the
meeting if there are any changes to the above pending items.

Table of Contents
Page 258 of 273
f. Once the meeting Pod has reconciled the meeting notes and posted in Teams that you can
write your letter, review the “Committee Review Submitted” information under the CR
history.
2. If the CR is pended or deferred:
i. Write a letter to the study team, using the appropriate template. Make sure you include all the
required changes noted in the “committee review submitted” information. Click “Send letter”
to end this part of the process.
ii. Follow up with the study team if a response is not received in a timely fashion. If a submission
is in requested changes for more than 60 days with no answer from the study team, the analyst
should withdraw it. The analyst needs to document their efforts in contacting the study team
and email them before withdrawing the study.
iii. Once the study team has submitted the requested changes, review materials to assess that the
changes have been made.
1. If Deferred issues have been addressed, schedule the Deferred CR to be reviewed by the
same committee that conducted the initial review of the CR application (i.e. if CMTE B1
reviewed the CR in January meeting and deferred it, send to the next available meeting for
CMTE B1). If the Deferred issues are approved at full board, you may issue the approval
letter and release updated consent/HIPAA documents.
2. If pending issues were addressed, email the meeting chair with a link to the study so they
can verify the changes were addressed.
Ask them to do not make any changes in
the submissions. The vice-chair may
document their approval via a private
logged comment, or they can email you
back with that response.
3. Once you receive the approval, click on
“Review Required Modifications” to
document the changes made are
adequate. The “approval date” would be
the date of the meeting. The “effective
date” is the date the vicechair confirmed
the changes were adequate and the study
can receive final approval.
4. Make sure you log a private comment
with the email communication from the
vice-chair if they did not log a private
comment.
Finalizing Submission
Note: Due to the Revised Common Rule, studies in data analysis only and in long term follow up (not
under FDA or DOJ regulations), or doing research with secondary data or biospecimens, that were

Table of Contents
Page 259 of 273
approved after 1/21/2019 or that transitioned to the Revised Common Rule, will not require continuing
review in the future. In the approval letter, add a reminder that Modifications, reportable new
information submissions, and closeout are still required. Also, if the status of the study changes
during the year, they need to submit a Modification (for example, if reopening accrual).
1. Go to page 25 of the Huron Staff Quick Reference
for technical steps for this part of the process.
2. Click “Finalize Documents”
3. Select the protocol and if the study is still enrolling, the consent form(s).
4. Click “Prepare Letter”.
5. Generate the appropriate letter. Remember to include the PI’s post-nominal (degree – MD, Ph.D.,
MPH, etc.), Principal Investigator (under name), and department name. If this information is not
clear in the submission, you can check the last approval letter, or you can search on the
Emory
Online Directory.
6. Review the letter to make sure all the information added by the system is correct including the
dates.
• If the study migrated to the 2018 Revised Common Rule, add a reminder that Modifications,
reportable new information submissions and a close out are still required. In addition, if the
status of the study changes during the year, they need to submit a Modification (for example, if
reopening accrual).
7. Delete template language that does not apply and put your name and title at the end of the letter.
8. Save a copy on your computer to upload a revision.
9. Click on the ellipses in the Draft letter space to upload the revision and click Ok.
10. Click “Send Letter”, review the letter and dates one more time, and click Ok.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
12/2/2016
Clarified how to determine expiration date; what to put in CR approval letter (removed some
bullets); referred to 30-day window SOP
5/5/2017
Clarified some steps in the process and added required language under FB approval letter
7/14/2017
Formatting and additional guidance on letter writing, comment logging.
5/17/2018
Revamp for readability
11/1/2018
Added location for the over enrollment log excel sheet (link does not work); added information about
when is OK to send a study to an expedited reviewer or to full board when finding expired research
training
1/18/2019
Adding information for Revised Common Rule. Clarifications and grammatical corrections.
8/31/2019
Updated customer service link.
2/11/2020
Updated CR to align with new eIRB
2/13/2020
Updated links to checklists
3/4/2020
Added that the pre-review note should indicate the enrollment numbers at the time to assign a study
to FB
3/13/2020
Updates in the information that needs to be added for CRs going to full board in the pre-review notes
3/23/2020
Updates in the revised common rule transition plan and addition of timelines to send to DR or FB.
4/14/2020
Updates in process for expedited review that were incorrect. More detail provided for contingency
review.
5/8/2020
Clarification on the steps after a contingency review changes are received after DR review.
6/14/2020
Updates to the Revised Common Rule transition process
8/21/2020
Instructions added when a study is transitioned to the revised common rule
12/11/2020
Adding additional information for the pre-review process for submissions going to FB
01/20/2021
Clarification of steps
2/25/2021
Adding that the submission can be withdrawn if no response from the team
5/5/2021
Changes in the contingency review process

Table of Contents
Page 260 of 273
SOP Title:
Continuing Review: Applying 30-day window
SOP
Study Management
Established:
10/14/2012
Last Revision:
2/11/2020
GLOSSARY
Review date: date on which expedited reviewer finally approved, or full board meeting occurred
Effective date/Approval date: first day of an approval period (for purposes of 30-day rule)
Expiration date: last day of an approval period (study expires at end of that day, at midnight).
Note: For stamping ICF/HIPAA documents, this will also apply if the study team requests an expiration
date in their documents.
SCOPE
All IRB staff may use the 30-day rule in the event of the following:
1. Study is no longer recruiting subjects, so they do not need restamped informed consent/HIPAA
documents and
2. Approval of the CR happens to occur during the same approval period as the year before (no need
to intentionally hold review until 30 day period arrives).
Scenario using the 30-day rule
• Current approval period: 7/2/2011 – 7/1/2012
• Renewal approved by the expedited reviewer on 6/26/2012 (within 30 days before 7/1/2012)
• New effective approval period: 7/2/2012 - 7/1/2013
PROCEDURE
• Step 1: After the designated reviewer has completed his/her review approving the study, click
on “submit designated review” to update the Approval Date and Effective Date.
• After finishing updating the dates, click on OK.
• Be sure to check the approval period granted by the reviewer/full board, if less than one year
• Make sure that the expiration date populated. Revised the letter as usual.
• Add the following language to the letter:
Because this study was reapproved [with conditions] within 30 days of its expiration date, is closed to
new enrollment, and thus consent forms no longer need to be stamped, we are applying guidance from
OHRP and FDA (see http://www.hhs.gov/ohrp/policy/continuingreview2010.html#section-g3 and
http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM294558.pdf) in order to retain
the approval and expiration anniversaries from the previous approval period.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
4/30/2015
Major overhaul, limits scope of when to use 30-day rule
11/1/2017
Revised language to mirror eIRB
1/31/2018
Date fix in example
2/11/2020
Updating to align to new eIRB system


Table of Contents
Page 262 of 273
SOP Title: Continuing Review: REs/PDs/Noncompliance and Monitor Reports
SOP Category:
Study Management
Established:
2/14/2012
Last Revision:
11/1/2017
PURPOSE: To explain what safety/compliance-related documents, submitted by the study team
with
a renewal application, should instead be submitted via the Reportable new information
submission (RE) form; and when such
documents should be removed from the Continuing Review
(CR) application.
SCOPE: Continuing Review submissions in eIRB – expedited and full board.
RESPONSIBILITIES:
•
IRB Analyst: Screen the Continuing Review (CR) application and review the
REs/PDs/Noncompliance and Monitor Reports per the
guidelines below
•
Team Q Lead (QTL): Receive alerts when study teams have been asked to submit REs,
and
follow up to ensure they are submitted as applicable
•
IRB Director: Escalate request for REs with study team as needed
RATIONALE: Leaving reports in the renewal application opens the IRB to liability when the IRB
member(s) do not adequately review those documents to determine whether they represent
unanticipated problems (UPs) or serious or continuing noncompliance.
OVERVIEW
Summary of Reporting Timelines
Unanticipated problems
Promptly reportable
Reportable protocol deviations
Promptly reportable
Internal deaths considered related
Promptly reportable
Internal deaths considered unrelated
Periodically reportable (i.e. at CR)
Internal related events (not UPs)
Periodically reportable (i.e. at CR)
External* SAEs that are not UPs
Never reportable to the IRB
External* deaths not related or UPs
Never reportable to the IRB
Noncompliance
Promptly reportable
*Remember that external events under the oversight of an Emory S-I should be assessed as internal
events.
PROCEDURE: IRB Analyst screening a CR submission should open every attachment to make sure that only
items that are properly reported at CR are remain in the submission, and that they are in the correct form. Any
items that required prompt reporting must be reported via a reportable new information submission, if
not previously reported. Other attachments should be removed from
the submission or modified to
comply with IRB requirements of reports at CR.
What should be reported in the CR?
•
The study team should submit periodic reportable new information submissions in a summary.

Table of Contents
Page 263 of 273
If not done, or the information provided was not clear, ask the study team to fill out our
summary form (if the
information they provided is complete, this is not needed). Use our
template at:
http://www.irb.emory.edu/documents/Sample_RE_summary.xls.
•
If the study team indicated that events did occur (by checking ‘yes’ to Questions 3-6 on page 1)
and/or submitted
descriptions of individual events in the last approval period, check to see if
the summary of events contains the following:
o
A list of previously reported UPs
o
A list of internal, expected (anticipated) and related events from the last approval
period
o
A list of internal, unrelated deaths
o
List of expected and related events and/or unrelated deaths for any external site under the
oversight of an S-I
•
For any UPs, make sure that the UPs listed were previously
reported to the IRB via a Reportable
new information submission. In order to check for previously reported items, go to the “AM-CR-
RE-Term” tab in the study record
•
If the IRB Analyst is unclear whether events are reportable (promptly vs. periodical) or not,
please consult Team Q and the
website guidance.
What should be removed from the CR and submitted as a RNI (unless the event is merely part of a
summary of events that were
already reported via RE)?
•
483 reports (FDA inspection findings)
•
Any event/death/protocol deviation/noncompliance report that met the criteria for being
promptly reportable:
o
Unanticipated problems (including external deaths that are considered a UP)
o
Reportable protocol deviations
o
Internal deaths considered related to the research
o
Noncompliance
o
Any report that indicates an increase in risk for the rights, welfare or safety of subjects
•
If the study team submits a spreadsheet with mixed information (SAEs reportable at CR
and events that are not reportable), ask them to remove the events that are not required
and submit in a reportable new information submission.
Procedure for handling CRs with items that require a RE:
•
If the study is close to expiration:
o
Remove the promptly reportable items from the submission
o
Email the study team and
CC the QTL, asking them to submit in an RNI within 2
weeks
o
Log a private comment in the CR documenting the email.
o
Process the rest of the CR as usual
•
If the study is not close to expiration (e.g. more than three weeks for expedited, or longer for full
board studies):
o
Send the CR back to the study team asking them to submit the list in a RNI instead of in the CR,
and to submit the CR back after they have submitted
their RE
o
Email QTL to alert Q
o
Log a private comment in the CR documenting email to QTL
o
If the study team does not respond and the study may expire, check with the
QTL for next

Table of Contents
Page 264 of 273
steps
What should be removed from the CR with notification by logged comment to study team, but no RNI
required?
•
Any items that are not periodically or promptly reportable
•
Minor protocol deviations (per the study team)
o
If the study team
needs an acknowledgment for protocol deviations, they need to
submit a separate RNI form.
•
External deaths: if they are not UPs (per the study team)
•
Site Monitor’s Report (not to be confused with a DMC/DSMB report)
o
Monitoring reports should be submitted to CTAC, not IRB. Email the monitoring
report(s) to CTAC/Stephanie deRijke at smickle@emory.edu
or
ctcompliance@emory.edu, CC the study team.
o
Remove the monitoring report and log a comment to study team saying you
removed the monitoring report, and have emailed it to CTAC
•
Example language: “Dear study team, we have removed document(s) X from your submission, as
it is not a document required for this review. If you need an acknowledgment, please submit
document X as a reportable new information submission”
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
10/3/2014
Added step of removing monitor report from submission; updated links
8/12/2015
Revamped this SOP to include what to report and what not to report at CR, as well as procedures to
deal with events
11/1/2017
Administrative updates and suggested language for notifying teams of information removal of
information
10/12/2020
Updated links, corrected typos

Table of Contents
Page 265 of 273
SOP Title:
Continuing Review: Processing study staff noncompliance with CITI and Clinical
Research Training (formerly Key Concepts/Intro to CR) requirements
SOP Category:
Study Management
Established:
4/30/2015
Last Revision:
4/20/2017
PURPOSE
The purpose of this document is to outline required actions when IRB staff confirms missing or
incomplete training (Clinical Research Training [CRT] for investigators and coordinators) while processing
a continuing review. Currently, during screening for a continuing review, the IRB staff check CITI training
for all study team members, and check CRT only for researchers who were added within the past
approval period via the IRB website's staff-change form.
SCOPE
The SOP applies to all studies approved by the Emory IRB.
RESPONSIBILITIES
• IRB analyst – to review CR information and confirm the completion of training requirements.
The IRB analyst will forward NC issues to Team Q
• Team Q: - to review any Reportable new information submission that is required
PROCEDURE
• Researcher did not have current CRT: the CR should not be sent to an expedited reviewer while
the training is being completed. The study can be placed on a FB committee agenda but this
issue should be included in the omnibus form under pending items. The study team could also
remove the person with the expired CITI from the submission (if not critical to study and PI
agrees). This person may be added back after they certify. Report the person to Bridget Strong
at BSTRONG@emory.edu
. Have the study submit a RE.
• Researcher is found to have never had CITI training from Emory or any other institution: Have the
team submit a reportable new information submission. When submitting the event, study team
should be asked to clarify whether the staff in question engaged in human subjects’ research,
and, if the online Staff Change web form was used, to identify who added the uncertified staff
person to the study. Follow additional procedures as indicated in the Training Verification SOP.
•
Researcher was added via web-form during the past approval period, but does not have current
CRT or/and current Biomedical or Socio-behavioral CITI training: Have the team submit a
reportable new information submission. When submitting the event, the study team should be asked
to clarify whether the staff in question is/was engaged in human subjects’ research.
• Researcher had Emory CITI certification but it was expired: the CR should not be sent to an
expedited reviewer while the training is being completed. The study can be placed on a FB
committee agenda but this issue should be included in the omnibus form under pending items.
The study team could also remove the person with the expired CITI from the submission (if not
critical to study and PI agrees). Person may be added back after they certify. A reportable new
information submission is not required.

Table of Contents
Page 266 of 273
• Researcher never had Emory CITI certification, but was CITI certified at another institution: the
CR should not be sent to an expedited reviewer while the training is being completed. The study
can be placed on a FB committee agenda but this issue should be included in the omnibus form
under pending items. The study team could also remove the person with the expired CITI from
the submission (if not critical to study and PI agrees). Person may be added back after they
certify. A reportable new information submission is not required.
• Researcher had the wrong Emory CITI training (Biomed/SHB): the CR should not be sent to an
expedited reviewer while the training is being completed. The study can be placed on a FB
committee agenda but this issue should be included in the omnibus form under pending items.
The study team could also remove the person with the expired CITI from the submission (if not
critical to study and PI agrees). Person may be added back after they certify. Review Step-by-
Step Guidance for Selecting Required Courses and Obtaining a CITI Account along with a
description of the research activities to determine if they really took the wrong course. No RNI
required in any case.
• Researcher had an incorrect Emory CITI training (GCP or RCR, and never took Biomed/SHB):
Have the team submit a reportable new information submission. When submitting the event,
study team should be asked to clarify whether the staff in question engaged in human subjects’
research and who added the staff to the study (if the online form was used). The CR should not
be sent to an expedited reviewer while the training is being completed. The study can be placed
on a FB committee agenda but this issue should be included in the omnibus form under pending
items. The study team could also remove the person with the expired CITI from the submission
not critical to study and PI agrees). Person may be added back after they certify.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
8/5/2015
Clarified that studies should not be sent to the expedited reviewer if training is not
complete. OK to send to FB but study will be pending until training is completed.
Added link for guidance on the website
9/16/2016
Updated terminology for the Clinical Research Training (formerly Key Concepts)
4/20/2017
Clarified what “correct” meant

Table of Contents
Page 267 of 273
CLOSE OUT
SOP Title:
Close-Out Process
SOP Category:
Study Management
Established:
7/21/2014
Last Revision:
07/06/2023
PURPOSE
To outline the processes for closing out a study in eIRB, including:
1. Close-outs initiated by a PI
2. Steps in eIRB to process the close-out
SCOPE
Applies to all IRB staff personnel responsible for processing close-out requests. For S-I studies go to SOP
entitled:
“Review of Studies Involving an Emory Sponsor/Sponsor-Investigator at Emory Holding an
IND/IDE”
PROCEDURE
NOTE: Closeout applications will start in the form of a Continuing Renewal Application.
1. Review the printer version of the renewal application. If all of the first 4 items in question 4 are
checked it indicates that the research is completed, and the study is ready to close out. Log a
comment to the study team stating that you have been assigned this closeout for review and that
you will be processing it.

Table of Contents
Page 268 of 273
2. Continue reviewing the submission for accuracy:
• Are the study numbers correct?
• Did they provide any DSMB reports, etc.? We will not require a summary. If applicable we will
need to see the latest DSMB report, unless the study has been in data analysis since the last
approval period. AA will review the DSMB document, and if it says anything other than no issues
or continue as planned, then consult w/an AD to determine if it needs to be reviewed by faculty.
o For closeout of S-I studies, look for the S-I closeout SOP.
• Have all requested reportable events been submitted? If applicable; this applies if the study was
still enrolling or doing research activities in the last approval period.
3. Once the submission is ready to be assigned to a reviewer (yourself). Click Submit Pre-Review. The
Pop-up window will allow you to capture all of the details of the study as well as make any relevant
notes.
4. Assign the study for review. Click on “Assign Designated Reviewer”. Analysts can process close-out
requests unless the study team is reporting a reportable event issue. If that is the case, the close-
out should be reviewed by a vice-chair. Contact a team Q member to assess if an RNI is needed.

Table of Contents
Page 269 of 273
You will need to complete the fields within the reviewer form:
1. * Determination:
Name
Related Worksheet
Approved HRP-314 - WORKSHEET - Criteria for Approval
2. * Risk level:
Greater than minimal risk
No greater than minimal risk
N/A
Clear
3. * Review level:
Name
Related Worksheet
Expedited HRP-313 - WORKSHEET - Expedited Review
Clear
4. * Indicate the categories: Select “Other”
5. * Rationale for continuing review requirement:
6. Dates: (Don’t worry about these pre-populated dates)

Table of Contents
Page 270 of 273
7. Notes:
Supporting documents: Not applicable
8. * I do NOT have a conflicting interest:
9. * Are you ready to submit this review?
Yes No Clear
5. Click “ok”
6. Prepare the letter. From the drop-down menu select the “IRB Study Closure” template letter if the
study is being administratively closed by the IRB. Select “PI Study Closure” template letter if the PI is
closing the study.
7. Check the letter that was generated. Add your signature. Re-upload the letter and click Ok. After
that step, you will be back in the submission. Click on “Send”. A new window will open. Check the
letter one last time and then click on OK.


Table of Contents
Page 272 of 273
SOP Title:
Informing Teams of Study Closure
SOP Category:
Study Management
Established:
7/21/2014
Last Revision:
11/3/2020
PURPOSE
The purpose of this document is to detail the process of informing study teams that their studies will be
closed out.
SCOPE
The SOP applies to expired studies approved by the Emory IRB.
RESPONSIBILITIES
• IRB Staff Leadership- have “IRB Director” role.
• IRB analyst: Informs the staff leadership to close a study using the “close Study (Admin)” function.
PROCEDURE
Before study expiration
• If a study is identified as one that may expire in the next 2 weeks, the IRB analyst will log the
following comment and sent via email:
Dear team,
Your study (IRB#, Short name-INSERT LINK) expires on DATE. Please submit a continuing review
application as soon as possible, as we need time to review it before approval. If the study has an
approval lapse, you will be required to submit a reportable event for noncompliance. Please, refer to
our timelines
for review of studies for additional information.
Alternatively, if all human subject research activities are complete, please Indicate in your submission
that you would like to close the study.
If your study is expired more than two weeks and no action is taken, your study may be closed
administratively.
If you have any questions, please contact your analyst directly.
Best regards,
YOUR NAME
Less than 90 Days Expired
• If the study expired less than 90 days with a continuing review in pre-submission:
o Reach out to the study team via email (copying analyst) and logged comment, telling them they
have 5 business days to submit the (CR as appropriate) or explain why more time is needed.
Here is a suggested text for when a CR has been created but not submitted:
Dear study team,
This study expired on [DATE]. Let us know if you have conducted any research during the
approval lapse. If so, please submit a reportable event. If this study is FDA regulated, you

Table of Contents
Page 273 of 273
will need to submit a reportable event even if research activities were not conducted during
the lapse. If you plan to continue with this research study, please submit a continuing review
application (you created one on [DATE] that has not been submitted yet). If you are no
longer conducting research activities, please submit a close out submission. Stop all
research, including data analysis until you secure reapproval. If we do not receive a
continuing review application, we will close out this study administratively in the next two
weeks. If you have questions or concerns, email or call your analyst: [insert analyst name,
email, number].
Here is a suggested text for when a CR to close out the study has been created but not
submitted:
Dear study team,
This study expired on [DATE]. Let us know if you have conducted any research during the
approval lapse. If so, please submit a reportable event. If this study is FDA regulated, you
will need to submit a reportable event even if research activities were not conducted during
the lapse. . If you are no longer conducting research activities, please submit a close out
submission (you created one on [DATE] that has not been submitted yet). Stop all research,
including data analysis until you secure reapproval. If we do not receive a continuing review
application, we will close out this study administratively in the next two weeks. If you have
questions or concerns, email or call your analyst: [insert analyst name, email, number].
• If the study expired less than 90 days without a CR created:
o Reach out to study team via email, copying the analyst-owner of the study (if not you), and as a
logged comment in eIRB, letting them know they have 5 business days to submit a continuing
review application.
• If an IRB analyst has a study that has been expired more than 3 months, a staff member with
“Director” role can close the study using the “Close Study (Admin)” option.
If a study team wants to reopen the study
The study team should submit a new submission, that would include using our current templates and
checklists and receive approval from any applicable ancillary reviewer
If a study was closed by mistake
Contact a Director. In general, we will have to ask the team to create a new submission and report the
issue to other ORA offices if applicable. The study team will be able to submit their current study
documents, and we will just move the study to approved and put today’s date as the expiration date (if
the system allows, it is preferable to keep the approval/expiration dates of the closed study). If the
study lapsed, the study team will need to submit a new CR application.
LOG OF SIGNIFICANT CHANGES
DATE
SUMMARY OF SIGNIFICANT CHANGES
2/11/2020
Removed items that cannot be executed in the new IRB system.
11/3/2020
Added additional information about the process
